![[软件使用 3] 使用MACS2分析ChIP-seq数据,快速入门!](data:image/svg+xml;utf8,<svg xmlns='http://www.w3.org/2000/svg'></svg>)
[软件使用 3] 使用MACS2分析ChIP-seq数据,快速入门!

本文主要包括:
- 一、软件下载和基本概念
- 二、数据下载与比对(sra-tools, fastqc, multiqc, trim_galore, bowtie2, samtools)
- 三、数据分析与画图 (masc2, igv, R/Python)
一、软件下载与基本概念
首先,我们将所需软件下载到Linux,同时在等待软件安装时了解一些基本概念。
1.1 软件下载
推荐使用“Anaconda”安装和管理软件,具体可以查看这篇文章:
如果你已经安装好“Anaconda”,那么接下来使用它下载好这些软件:
conda install -c bioconda sra-tools -y # sra-tools用于下载数据
conda install -c bioconda fastqc -y # fastqc查看测序数据质量
conda install -c bioconda multiqc -y # multiqc可以合并多个fastqc的结果到一个html
conda install -c bioconda trim_galore -y # trim_galore去除接头和低质量的 reads
conda install -c bioconda bowtie2 -y # bowte2是比对软件
conda install -c bioconda samtools -y # samtools 处理sam和bam文件
conda install -c bioconda bedtools -y # bedtools 处理 bed 文件
conda install -c bioconda macs2 -y # macs2 用于Peak Calling
conda install -c bioconda igvtools -y # IGV的命令行工具
conda install ucsc-wigtobigwig -y # wig 转 bigwig
conda install ucsc-bedgraphtobigwig -y # bedgraph 转 bigwig
conda install -c bioconda ucsc-bedclip -y # bedclip 删除bed文件中不在染色体上的行
# -y 表示安装时不询问IGV是Broad Institute(伯德研究所)开发的软件,可以对基因组比对结果进行可视化。IGV下载链接:Downloads | Integrative Genomics Viewer 。
另外,也要确认本地是否安装X manager,并且在X Shell设置使用X11转发功能。具体操作可以查看 Xshell使用X11转发功能 | Xmanager中文官网。这一步是为了在Linux系统下打开IGV。
这些软件的具体使用方法和需要的注意细节将在下文中说明。
1.2 基本概念
- ChIP-seq
ChIP-seq,全名Chromatin Immunoprecipitation,中文名“染色体免疫共沉淀技术”。主要用来研究蛋白质和DNA的相互作用。具体来说,就是确定特定蛋白是否结合特定基因组区域,或者基因组上是否有与组蛋白修饰相关的特定位点。
ChIP-seq的大致流程:
1. 将蛋白交联到DNA上,其实就是让蛋白和DNA结合,让我们可以找到互作位点;
2. 使用超声波或酶剪切DNA链;
3. 加上结合有特定抗体的磁珠用于免疫沉淀靶蛋白;
4. 解除蛋白交联,纯化所需DNA;
5. 将纯化后的DNA进行高通量测序。
- Reads
测序仪得到的原始图像文件,经过碱基识别和误差过滤后得到的可以用于分析的原始测序片段为 Reads,一般以 fastq 格式储存。
- Phred quality score
Phred 最初是一个从测序仪中产生的荧光记录数据中识别碱基的程序。
在早期的荧光染料测序中,每次发生碱基合成时会释放出荧光信号,该信号被CCD图像传感器捕获。记录下荧光信号的峰值,生成一个实时的轨迹数据(chromatogram)。因为不同的碱基用不用的颜色标记,检测这些峰值即可判断出对应的碱基。但由于这些信号的波峰、密度、形状和位置等是不连续或模糊的,有时很难根据波峰判断出正确的碱基。
Phred 计算许多与波峰大小和分辨率相关的参数,根据这些参数,从一个巨大的查询表中找出碱基质量得分(不同的测序试剂和机器用不同的查询表)。为了节约磁盘空间,质量得分(可能占用两个字符)按一定规则(Phred+33或Phred+64)被转换为单个字符表示。
了解更多:Fastq 格式说明 & (Phred33 or Phred64).
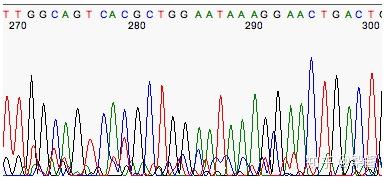
- 测序数据过滤
测序下机的原始数据(raw reads),包含带接头和低质量的 reads。为保证后续分析的准确性,必须对原始数据进行过滤,得到 clean reads。后续的分析都是基于 clean reads。
- Peak Calling
Peak Calling,利用计算的方法找到ChIP-seq或ATAC-seq中 reads 富集的基因组区域。
- 软件使用的小技巧
软件的具体使用不用死记硬背,直接上网查询软件的帮助文档,并将每次使用的代码保存好做好注释。下次再看自己的代码注释即可~
养成注释好习惯,造福你我他。
- ChIP-Seq 常用分析流程
分析流程大同小异。
Tips:可以看些综述、博士论文、公司的分析报告让自己更好的了解ChIP-seq背景知识、发展脉络以及应用方向。


二、数据下载与比对
2.1 数据下载
2.1.1 使用 prefetch 下载数据
prefetch srrID #单个文件下载
prefetch --option-list srrID_list.txt # 批量下载,将需要下载的文件放在一个txt文件不推荐使用FTP下载。因为NCBI数据库更新,部分文件已经没有FTP链接,无法下载。
但是使用NCBI提供的SRA-tools工具中的prefetch,会因为网络原因(你懂的)经常中断,只能重头下载。
怎么办?
可以用for循环实现简单的“断点重新下载”:
for i in $(seq 1 100)
do
prefetch --option-list srrID_list.txt
done如果文件不多,只有二三十多个,基本上挂一晚上就能下载完。
2.1.2 使用 Fasterq-dump 将sra文件转为fastq文件
fasterq-dump -e 10 --split-3 SRRxxx.sra # -e 线程数注:也可以使用 aspera 直接下载 fastq 文件。
2.2 测序质量控制
2.2.1 使用 FastQC 做质控:
软件的具体用法,可以在命令行中输入 “fastqc -h” 查看。
其他软件也可以通过输入软件名加"-h"查看用法。
fastqc [-o output dir] [--(no)extract] [-f fastq|bam|sam]
[-c contaminant file] seqfile1 .. seqfileNFastQC 具体运行代码:
# 查看单个文件的测序质量
fastqc -o ../fastqc -f fastq -t 10 SRRxxx.fastq
# 查看多个文件的测序质量
for i in *.fastq
do
fastqc -o ../fastqc -f fastq -t 10 $i
done如何查看 FastQC 结果:孟浩巍:20160410 测序分析——使用 FastQC 做质控
2.2.2 使用 MultiQC 将多个 FastQC 的结果汇集到一个文件:
multiqc ../fastqc -n SRAxxx -o ../multiqcResults/
# multiqc 后面直接跟上 fastqc 结果所在的文件夹
# -n 输出结果的文件名前缀
# -o 设置输出结果的文件夹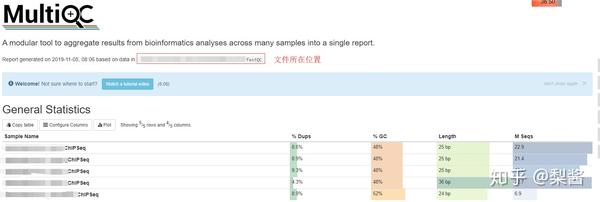

查看 Multiqc manual 了解更多。
2.2.3 使用 trim_galore 进行数据清洗
Trim Galore 是对 FastQC 和 Cutadapt 的包装,适用于所有高通量测序。
Trim Galore! is a wrapper script to automate quality and adapter trimming as well as quality control, with some added functionality to remove biased methylation positions for RRBS sequence files (for directional, non-directional (or paired-end) sequencing).
ls ~/chipseq/raw/*.gz | while read id;
do
nohup trim_galore -j 10 -q 20 --phred33 --length 25 -e 0.1 --stringency 4 -o ~/chipseq/clean $id 1>trim_galore.log &
done
# 出错时查看 trim_galore.log 可能是参数等输错了。
# -j 线程
# -q -quality<int> 设定phred quality阈值。默认20(99%的read质量),如果测序深度较深,可以设定25
# --phred33 可以选择-phred33或者-phred64,表示测序平台使用的Phred quality score
# --length 设定输出reads长度阈值,小于设定值会被抛弃
# --fastqc 同时做质控
# --fastqc_args 可以设置 fastqc 的参数2.3 比对
首先了解下我们使用的 Bowtie2 软件,它是什么?有什么用处?
Bowtie2 是一个超级快速、省内存的将短序列比对至模板基因组的工具。它尤其擅长于将50~100个核苷酸大小的短序列比对到基因组上。
Bowtie2 使用 Burrows-Wheeler Transform (BWT) 算法给基因组建立 FM 索引,这种方法内存占用空间小,便于进行快速比对。Bowtie 2 支持 gapped, local paired-end 比对模式,并且可以使用多个处理器让比对速度更快。
Bowtie 2 输出 SAM 格式的比对结果,从而便于和其他使用SAM格式的软件串联起来(e.g. SAMtools, GATK) . Bowtie 2 在 GPLv3 许可证下发布, 支持Windows, Mac OS X 和 Linux 下的命令行操作.
Bowtie 2 通常是比较基因组学研究的第一步, 包括 variation calling, ChIP-seq, RNA-seq, BS-seq. Bowtie 2 和 Bowtie (这里也可以称为 "Bowtie 1" ) 被整合到许多工具中, 其中一些列在这里。
参考文献:Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012 Mar 4;9(4):357-9. doi: 10.1038/nmeth.1923.
当然,你也可以选择其他比对软件,如BWA。这里我们仅介绍Bowtie2。
2.3.1使用Bowtie2建立参考基因组的index
不建议自己建立,太花时间,可以直接在 Bowtie2 网站下载。
但是如果 Bowtie2 网站上没有提供想要的 index,就要到 illumina iGenomes 网站 下载对应基因组建立 index。
如何建立?这里用 hg38 举例:
bowtie2-built --threads 10 -f hg38.fa hg38
# -p/--threads 线程数
# -f 输入fasta文件
# 最后一个hg38是输出文件的前缀
nohup bowtie2-built --threads 10 -f hg38.fa hg38 & # 建议用nohup挂到后台2.3.2 将fastq文件比对到基因组上
for i in *fastq
do
bowtie2 -p 5 -q $i -x /bowtie2_index/hg38_UCSC/hg38 -S /analysis/bowtie2/$i.sam
done
# -p/--threads 线程数
# -q 输入fastq文件
# -x index所在路径
# -S 输出sam文件了解更多可参考官方文档:bowtie2 manual
2.3.3 使用samtools进行排序
samtools sort SRRxxx.sam -o SRRxxx.sam.sorted.bam这个 BAM 文件就是后面 MACS2 的输入文件。
了解更多可参考官方文档:samtools manual
2.4 使用IGV可视化比对结果
2.4.1 打开Xmanager,在命令行输入:
xterm &2.4.2 运行IGV软件,在 Xmanager 中输入:
cd ./softwares/IGV_Linux_2.7.2 # 切换到igv.sh所在目录
./igv.sh # 打开igv2.4.3 载入参考基因组数据到IGV:
基因组的fasta文件可以在UCSC下载,然后用 samtools 建立索引。
#对参考基因组建索引,如hg38
samtools faidx hg38上传基因组,软件不支持压缩格式:Genomes->Load Genome from File->选择genome.fa并打开
2.4.4 载入sort后的BAM文件到IGV:
#对比对结果建立索引
samtools faidx SRRxxx.sam.sorted.bam上传sort后的BAM文件:File->Load from File->选择sample.sort.bam并打开。
注意:如果BAM文件太大,可以使用 igvtools 将BAM转为TDF文件再可视化:
igvtools count -z 5 -w 25 *.bam *.bam.tdf hg38但是TDF文件只能反映基因组每个区域的测序深度,无法看到具体的比对情况,适合用来检查已经找到的Peak或者CNV。~o( =∩ω∩= )m
2.5 补充:使用UCSC可视化比对结果
当然,你也可以选择不用 IGV 而用 UCSC 可视化基因组的比对结果,因为UCSC提供更多选项,并且图形更美观。
但是 USCS 需要我们提供 BIGWIG 文件,我们现在手里只有 sort 后的 BAM 文件。
问题1:BAM和BIGWIG格式是什么?
- BAM 文件是 SAM 的二进制转换版。
- BIGWIG 是 wig 或 bedGraph 的二进制版,存放区间的坐标轴信息和相关计分(score),主要用于在基因组浏览器上查看数据的连续密度图。可用UCSC的工具 wigToBigWig 程序将 wig 转换为 bigwig。
wig 和 Bedgraph 格式是什么?这里我们顺带了解下。
USCS 帮助文档称 bedgraph 和 wig 格式是已经过时的基因组浏览器图形轨展示格式。Bedraph展示稀松型数据,wig展示连续性数据。目前推荐使用 bigWig/bigBed 这两种格式取代前两者。
更多关于格式的资料,参考:[生信资料 3] 生物信息学常见数据格式,汇总!
问题2:为什么可视化要用 bigWig?BAM文件不行吗?
主要是因为 BAM 文件比较大,直接用于展示时对服务器性能要求较大,加载可能会非常慢。因此在GEO上一般只会提供bigWig文件,便于研究人员下载和查看。 如果真的对数据感兴趣,则可以下载原始数据自行分析。
问题3:怎么将手头的 BAM 转为UCSC可视化需要的 BIGWIG 格式?
工具一: Bam2bigwig, a tool to convert bam files into bigwig for UCSC Genome Browser
用法:
chmod 775 bam2bigwig
./bam2bigwig file.bam
# 但是要注意代码中使用的是hg19,要自行按需更改为其他版本
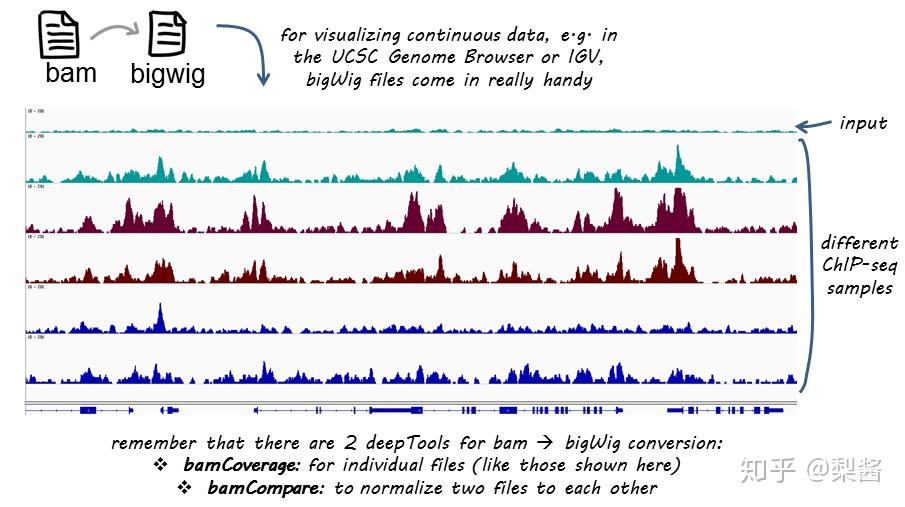
工具二(推荐):deeptools bamCoverage
bamCoverage -e 170 -bs 10 -b test.bam -o test.bw 这里的参数-e/--extendReads拓展了原来的read长度,-bs/--binSize 设置分箱的大小。
可以自行搜索 deeptools 了解更多参数。
现在你就有了Bigwig文件啦~ 将该文件上传到UCSC,就能进行可视化:
打开UCSC---Tables---My Data---Custom Tracks

了解更多可参考:使用UCSC和IGV查看reads在基因组上分布情况. 熊朝亮. 科学网。
三、数据分析与画图
3.1 Peak Calling
MACS, Model-based Analysis of ChIP-seq. 主要用来寻找 peak。
MACS 软件由哈佛大学 X Shirley Liu 实验室开发,该实验室侧重于基因调控机制的生物信息和计算生物学研究。
建议在看下文前,先查看 taoliu/MACS/Manual 了解下 MACS2 的基本用法。
3.1.1 使用 MACS2 进行Peak Calling
MACS2 callpeak 根据有无 control 数据采用了不同的计算方法,但都是依据泊松分布。
具体原理可以查看原文献:https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2592715/ ,或者别人的解读:https://blog.csdn.net/BoringFantasy/article/details/80622699。
计算公式:λ_{local} = max(λ_{B}G, [λ_{1}k,] λ_{5}k, λ_{10}k)
MACS2使用 \lambda_{local} 计算 peak 的 p 值,没有 control 则没有 \lambda_{1k} 。
输入的数据为 2.3.3 samtools sort 后的文件。
#有control数据
for i in *.bam
do
macs2 callpeak -f BAM -c control.bam -t $i -n $i -g hs --outdir ../macs2/ --bdg -q 0.05
done
#没有control数据
for i in *.bam
do
macs2 callpeak -f BAM -t $i -n $i -g hs --outdir ../macs2/ --bdg -q 0.05
done
# -f 指定输入文件的格式,如SAM、BAM、BED等
# -c 对照组,可以接多个数据,空格分隔。
# -t 实验组,ChIP-seq数据。可以接多个数据,空格分隔。
# -n 输出文件名的前缀
# -g 有效基因组大小。软件有几个默认值,如hs指human
# --outdir 输出结果的存储路径
# --bdg 输出 bedgraph 格式的文件
# -q FDR阈值, 默认为0.05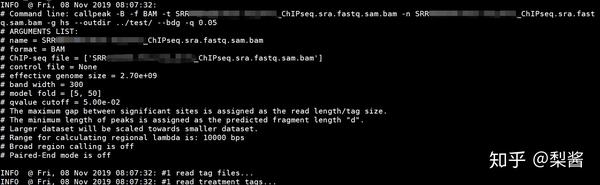
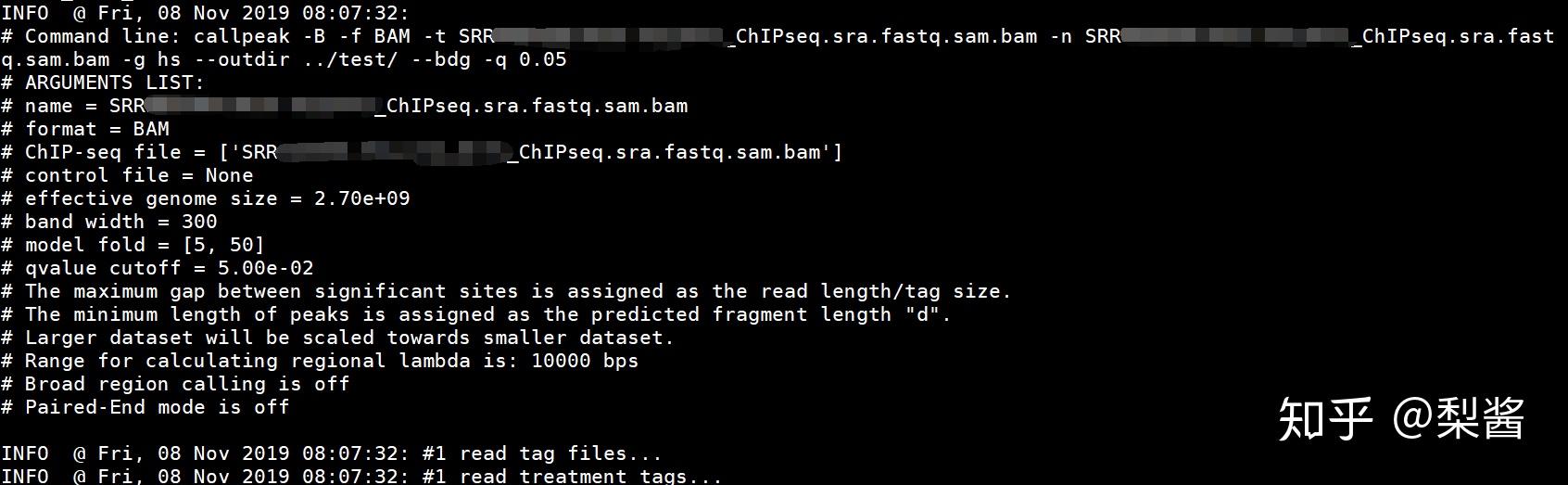
注意:使用 MACS2 call peak 时,有人可能会报错 “IndexError: list index out of range”,检查一下是不是因为你没有sort sam。
MACS2 得到的结果(taoliu/MACS/Manual 有具体讲解):


# NAME 是使用 -n 设置的输出文件前缀
1. NAME_control_lambda.bdg 和 NAME_treat_pileup.bdg
# bdg即为bedGraph格式,可以导入UCSC或者转换为bigwig格式。
# 上文2.5有这两种格式的说明。
# 两个bdg文件:
- treat_pileup
- control_lambda
2. NAME_model.r # R代码
3. NAME_peaks.narrowPeak # BED 6+4 格式
4. NAME_peaks.xls # 包含 peak 信息的 xls 文件,注意 chrStart 是1-based
5. NAME_peaks_summits.bed
# BED 格式,包含peak的summits位置,第5列是-log10pvalue。
# 如果想找motif,推荐使用此文件。 其中需要注意,NAME_model.r 以 R 代码的形式保存了“双峰模型”。
在命令行中输入:
Rscript NAME_model.r 会得到名为 NAME_model.pdf 的文件↓:

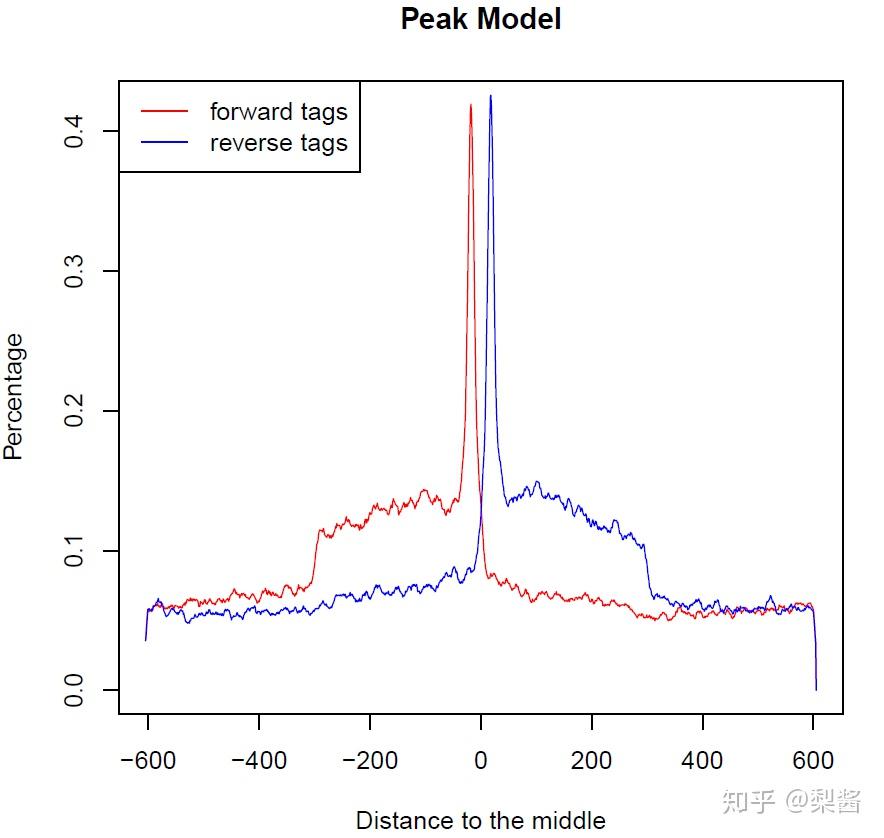
图中的‘双峰模型’是什么?为什么要建立它?有什么用?
首先我们要记住一点——所有数据分析的流程都基于实验原理。
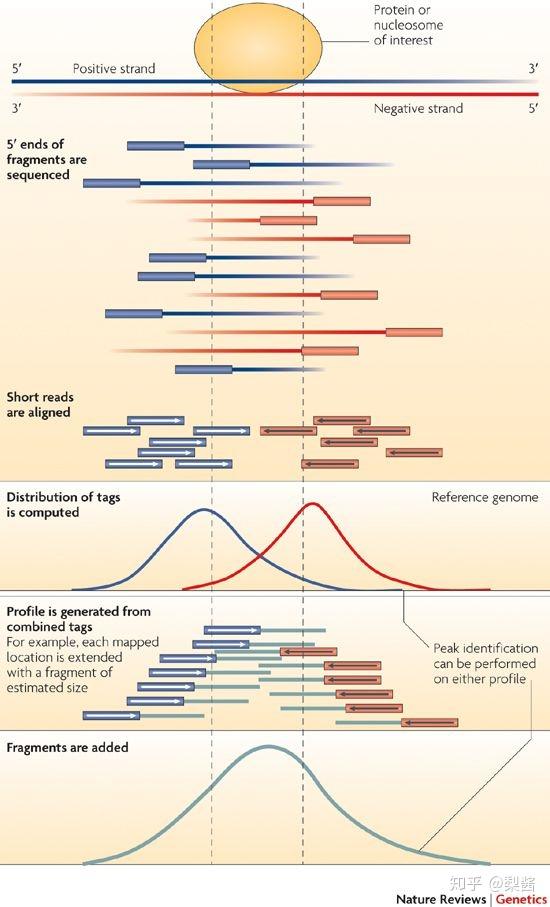
做 ChIP-seq,就是为了确认蛋白-DNA结合位点。测序获得的 read 只是跟随着蛋白一起沉淀下来的 DNA 片段的末端,并不是真实的 DNA 和蛋白结合的位置。怎么找到蛋白和DNA真正的结合位置?
- 单端测序,对于测序的同一片段,reads 在正负链上的分布是平均的,并且正负链的 reads 之间会有一个间距。近似的形成“双峰”。因此,把来自于同一片段位于不同链上的 reads 相向移动,就能有效地降低信号中的噪音,找到结合的位置。
- 而对 双端测序 而言,它本身测的就是文库的两端,因此不用建立模型和偏倚。我们只需要对 MACS 设置参数 --nodel 就能略过双峰模型建立的过程。
如下图:

参考:
- Model-based Analysis of ChIP-Seq (MACS). Y Zhang, T Liu et.al. Genome Biol. 2008; 9(9): R137.
- ChIP–seq: advantages and challenges of a maturing technology. P Park. Nature Reviews Genetics. 2009; volume 10: 669–680.
- https://www.plob.org/article/7227.html
3.1.2 Peak 可视化
- 方法1 使用 IGV 查看 BedGraph 文件
前面已经上传过基因组文件了,不再赘述。如果没有上传请查看2.4。
这里直接上传 bg 文件:File->Load from File->选择sample.bg并打开。
2. 方法2 使用UCSC 查看 BigWig 文件
上面(见2.5)我们是将 BAM 转为 BigWig,这里是将 MACS2 输出的 BedGraph 文件转为 Bigwig 文件,然后通过 UCSC 查看。
注意:这种方法比直接导入 IGV 复杂,不推荐,不推荐,不推荐!
网上有很多种将 BedGraph 转为 Bigwig 的方法,这里仅提供一种作为参考~
首先,使用 macs2 bdgcmp 得到 FE 或者 logLR 转化后的文件 :
# 使用 macs2 bdgcmp 得到 FE 转化后的文件
# -m FE 计算富集倍数降低数据噪音
macs2 bdgcmp -t NAME_treat_pileup.bdg -c NAME_control_lambda.bdg --outdir ../test -o NAME_FE.bdg -m FE
# 使用 macs2 bdgcmp 得到 logLR 转化后的文件
# -p 为避免log的时候input值为0时出现error,赋值为0.00001
macs2 bdgcmp -t NAME_treat_pileup.bdg -c NAME_control_lambda.bdg --outdir ../test -o NAME_logLR.bdg -m logLR -p 0.00001然后,复制 MACS2 共一作者 Tao Liu 的代码 bdg2bw.sh 并用 "chmod+x" 更改文件权限。同时下载人 hg38 版的染色体长度文件并解压: hg38, chromInfo.txt.gz,重命名为hg38_UCSC.chromInfo.txt。其他物种可以自行在UCSC下载。
# bedgraph to bigwig
sh /scripts/bdg2bw.sh NAME_FE.bdg /index/hg38_UCSC.chromInfo.txt > NAME_FE.bw
sh /scripts/bdg2bw.sh NAME_logLR.bdg /index/hg38_UCSC.chromInfo.txt > NAME_logLR.bw发现报错“bedGraphToBigWig: error while loading shared libraries: libssl.so.1.0.0: cannot open shared object file: No such file or directory”。
这里显示没有libssl.so.1.0.0 ,安装好后就可以了~
最后会得到文件:
NAME_FE.bw
NAME_logLR.bw具体选用哪个文件,要结合实际情况。
查看 taoliu/MACS/Build Signal Track 了解更多。
3.1.3 将 NAME_peaks_summits.bed 比对到基因组,查看目的蛋白结合的位点
单独把这一点拿出来说,是因为想提醒你注意 macs2 call peak 得到的 bed 文件中,链方向为“.”(无方向)。


为什么是无方向?
因为构建双峰模型时,同时使用了正链和负链的文件。
怎么比对?
自行在后面加一列 “+”或者“0”即可。
HOMER's "findPeaks" tool will accept input from sequencing runs that are strand-specific. This is why the value is included in the file specifications. But the default when calling peaks is to use "BOTH", which can be interpreted as all peak calls being unstranded (merged). HOMER interprets these as a strand assignment of "0" (translates to +).
In BED files, a dot "." can be used successfully with many tools to represent a "NULL" value for strand (this defaults to "+" when used with some tools). I would give this a try first, but I am not certain if it will work since I haven't tried it. So give it a test. If it doesn't work, just use "+" for all entries in your BED file.
When converting BED to a Peak file (using the tool provided in the HOMER tool suite), the "+" will be converted to "0". This is a good idea to do, since some of the tools in this package only work with strand annotated with "0" or "1". The start coordinate value will also be modified to have a 1-based start, instead of the 0-based start used in BED format.
https://www.biostars.org/p/138407/
3.2 画图
接下来就是对 Peak 进行可视化,常用的有两种工具:
- R 包 ChIPseeker ,可以参考包开发者 Y叔的文章:CS3: peak注释 。
- deepTools,参考 deepTools manual 。
可视化可以做很多事,如:
- 绘制韦恩图,描述不同蛋白与结合基因的关系;
- 绘制不同蛋白在染色体结合位点与TSS的距离;
- 结合位置的热图;
- 不同染色体结合同一蛋白质的位点比较等。
话不多说,我们先来试试 ChIPseeker。deepTools 我之后会单独写一篇文章,在这里先挖个坑。
因为权限问题,我在第一步安装ChIPseeker就卡壳,还好最后解决了……
如果你也碰到安装问题,可以看下去;没有碰到,可以跳过。
在Windows下安装 ChIPseeker 时,使用 R 3.5 总是报错。尝试在 R GUI 输入以下命令升级到R 3.6:
library(installr)
updateR()还是报错:
Installation path not writeable, unable to update packages: boot, foreign, KernSmooth, mgcv, nlme, survival
Old packages: 'farver', 'RcppEigen'"
# 翻译:R使用bioconductor下载的包,被安装到了一个没有“写入”权限的文件夹中。搜索了很久,终于发现报错原因:
boot, foreign, KernSmooth, mgcv, nlme, survival 这几个包是最开始安装 R 时,软件自带的,随 R 软件一起安装在系统路径C:/Program Files/R/R/library。你安装 R 软件的时候,R 向你要了管理员的权限,所以你能把这些包安装在系统路径。
但是,在使用install.packages 更新这些包时,默认你没有管理员权限 ,因此不能打开C:/Program Files/R/R/library更新这些文件,而ChIPseeker又需要这些包才能运行,这样就会出错。
这个问题的关键在于,你想在系统路径安装包,却没有管理员权限。好比,你没有通往一间房间的钥匙,却想往这个房间放东西,肯定就会出问题。
解决了权限(钥匙)问题,就可以安装包(往房间里放东西)了。
解决方法:
步骤一:在R studio中查看R包安装的路径,一般会发现有两个路径。
#查看R包安装目录
.libPaths() 

步骤二:
- windows用户
- 可以通过右键属性,修改系统安装路径 C:/Program Files/R/R/library 的权限。这一步请自行百度~
- Linux用户
- 如果你是root用户,命令行下使用“chmod 777 文件路径”就可以修改;
- 不是root用户的话,那么找root 用户帮你更新。
- 如果不需要调用原本在root目录下的文件,可以使用 lib 指定安装路径。
# lib 指定安装路径
BiocManager::install('XML', lib="C:/Users/Documents/R/win-library/3.6")步骤三:重新安装出现报错的这些包:
#安装boot, foreign, KernSmooth, mgcv, nlme, survival包
install.packages(c("boot", "foreign", "KernSmooth", "mgcv", "nlme","survival", "farver", "RcppEigen"))

终于搞定……
这里要注意,R 3.6 下载 Bioconductor 的包要用 BiocManager 而不是 BiocLite 或 BiocInstaller。
# R 3.6.1 (2019-07-05)
# 用BiocManager下载
install.packages("BiocManager")
BiocManager::install("ChIPseeker")
BiocManager::install("Rsamtools")
BiocManager::install("rtracklayer")
BiocManager::install("GO.db")
BiocManager::install("DO.db")
#下载人的TxDb对象
BiocManager::install("org.Hs.eg.db")
BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene")
BiocManager::install("TxDb.Hsapiens.UCSC.hg38.knownGene")
# 要先加载TxBb等文件,再加载ChIPseeker
library("TxDb.Hsapiens.UCSC.hg19.knownGene")
library("TxDb.Hsapiens.UCSC.hg38.knownGene")
library("org.Hs.eg.db")
library("ChIPseeker")如果觉得有用记得点赞~
有问题或建议,请在评论区留言~
阅读更多文章:
参考:
- Question: Macs2 callpeak error: IndexError: list index out of range
- Home | Integrative Genomics Viewer
- 可视化工具之 IGV 使用方法 - Life·Intelligence - 博客园
- [Linux] IGV使用说明-懂客-dongcoder.com
- [Linux] IGV使用说明 - 萧飞IDO - 博客园
- bam文件的可视化(测序深度) | IGV
- samtools manual
- bowtie2 manual
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012 Mar 4;9(4):357-9. doi: 10.1038/nmeth.1923.
- Multiqc manual
- 第3篇:用MACS2软件call peaks
- ChIP-seq 数据分析. fengzhibifang. CSDN
- 使用UCSC和IGV查看reads在基因组上分布情况. 熊朝亮. 科学网.
- 如何使用MACS进行peak calling. 徐洲更hoptop.
- 如何使用deeptools处理BAM数据. 徐洲更hoptop
- ChIP-seq基础入门. 徐洲更hoptop. 简书
- xuzhougeng/R-ChIP-data-analysis. Github.
- 第四章-chip-seq数据分析. qiubio.com.
- 安装包时为什么会被区分安装在两个路径中,其中一个路径显示安装不成功。统计之都论坛.
- 改变R语言默认存储包的路径. faith默默. CSDN
- Fastq 格式说明 & (Phred33 or Phred64). 揭文才科学网博客.
- Trim Galore ——自动检测adapter的质控软件. 六六_ryx
