
三代测序入门:PacBio数据分析

本文章已同步发表于universebiologygirl微信公众号,欢迎关注!

目录
1. QC
1.1. 下机数据
1.2. 数据结构
1.3. 数据质量
2. 组装
2.1. 组装策略
2.1. de novo assembly 算法
3. 测序错误校正
1. QC
1.1. 下机数据


在analysis文件夹中,下机的数据被分割为三个文件进行存储
- 以bax.h5为后缀的是原始二进制文件;
- 以subreads.fasta / subreads.fastq为后缀的是经一级处理得到的标准格式的碱基文件;
- 以sts.csv / sts.xml为后缀的是记录测序过程中每个ZMW度量指标的统计文件
数据的命名:
m 140415_143853_42175_c100635972550000001823121909121417_s1_p0
└1┘ └─────2─────┘└──3──┘└───────────────4────────────────┘└5┘ └6┘1. m是movie的缩写;
2. 测序时间,格式为yymmdd_hhmmss;
3. 仪器编号;
4. SMRT Cell Barcode;
5和6无实际意义,一般是固定的
1.2. 数据结构
Pacbio 数据的文库模型是两端加接头的哑铃型结构,测序时会环绕着文库进行持续的进行,由此得到的测序片段称为 polymerase reads,即一条含接头的测序序列,其长度由反应酶的活性和上机时间决定。目前,采用最新的 P6-C4 酶,最长的读长可达到 60kb 以上。


polymerase reads 是需要进行一定的处理才能获得用于后续分析的。这个过程首先是去除低质量序列和接头序列:


处理后得到的序列称为 subreads,根据不同文库的插入片段长度,subreads 的类型也有所不同。
对长插入片段文库的测序基本是少于2 passes的(pass即环绕测序的次数),得到的reads也称为Continuous Long Reads (CLR),这样的reads测序错误率等同于原始的测序错误率。
而对于全长转录组或全长16s测序,构建的文库插入片段较短,测序会产生多个passes,这时会对多个reads进行一致性校正,得到一个唯一的read,也称为Circular Consensus Sequencing(CCS)Reads,这样的reads测序准确率会有显著的提升。
polymerase reads 与 subreads 是相对应的两个概念
Continuous Long Reads (CLR) 与 Circular Consensus Sequencing(CCS)Reads 是是相对应的两个概念1.3. 数据质量
不同于二代测序的碱基质量标准Q20/Q30,三代测序由于其随机分布的碱基错误率,其单碱基的准确性不能直接用于衡量数据质量。那么,怎么判断三代测序的数据好不好呢?
- 长度
长度短的测序数据不一定差(与文库大小有关),但差的数据长度一定短。在上游实验环节,最关键的影响因素是文库的构建。高质量的文库产出的数据长度长,质量好;而低质量的文库产出的数据长度短,质量差。


- 比例
需要关注的是两个比例:
一个是subreads与polymerase reads数据量的比例,比例过低反映测序过程中的低质量的序列较多;
一个是zmw孔载入的比例,根据孔中载入的DNA片段数分为P0、P1和P2。P1合理比例在40%-60%之间。上样浓度异常会导致P0或P2比例过高,有效数据量减少。需要注意的是P2比例过低时,可能存在P2转P1的情况,测序结果包含较多的嵌合型reads。

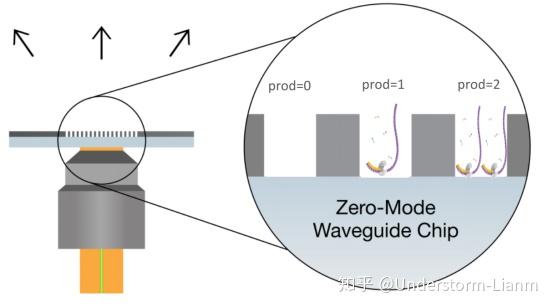
一张芯片上有15万个孔,其中只有大概三分之一有一个测序复合物(聚合酶+测序引物+测序模版),另外三分之一是空的,剩下的三分之一是有>2个以上的测序复合物产生的数据再接下来的分析中是要去掉
2. 组装
2.1. 组装策略
目前采用的组装策略:
PacBio-only de novo assembly :只使用 PacBio 产生的 long reads 进行拼接,在拼接之前要进行预处理,然后采用 Overlap-Layout-Consensus 算法进行拼接
Hybrid de novo assembly :结合 PacBio 的长reads 和 二代的短 reads
Gap filling :用二代的短reads(包括Pair-end和Mate-pair reads)拼接得到scaffod,然后用 PacBio 的长 reads 进行补洞
Scaffolding :用二代的短reads(包括Pair-end和Mate-pair reads)拼接得到 contigs / scaffod,用 PacBio 的长 reads 确定 contigs / scaffod 之间的位置关系

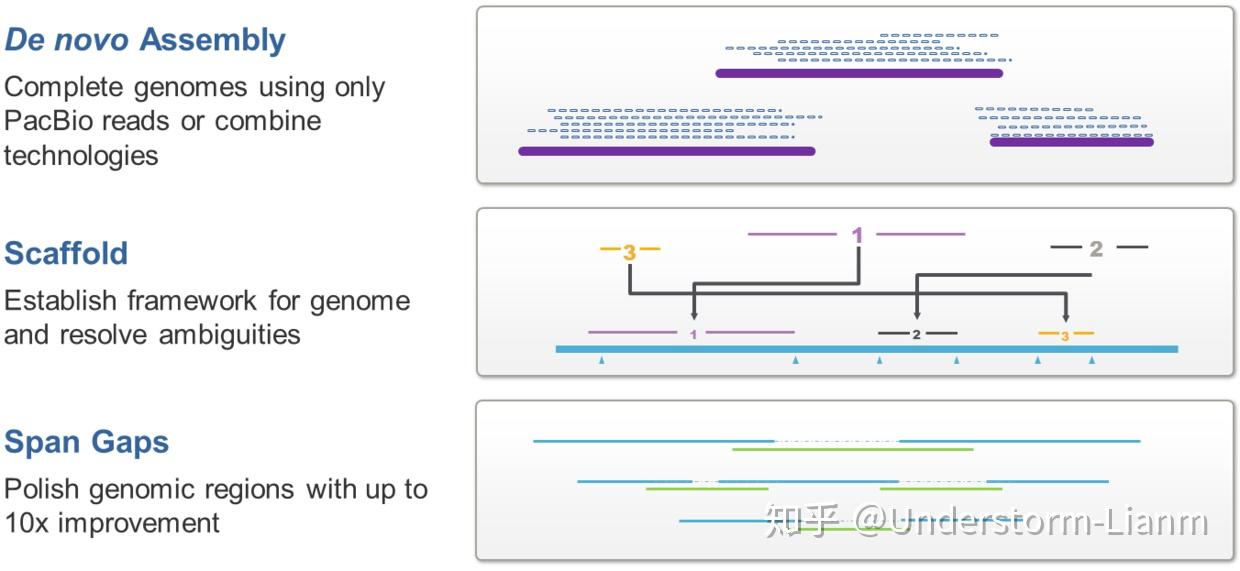
这四种组装策略并不是完全孤立的,在一个组装任务的不同阶段会用到不同的方法
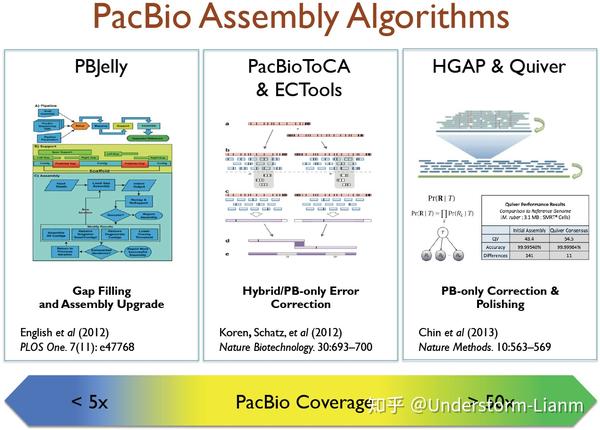
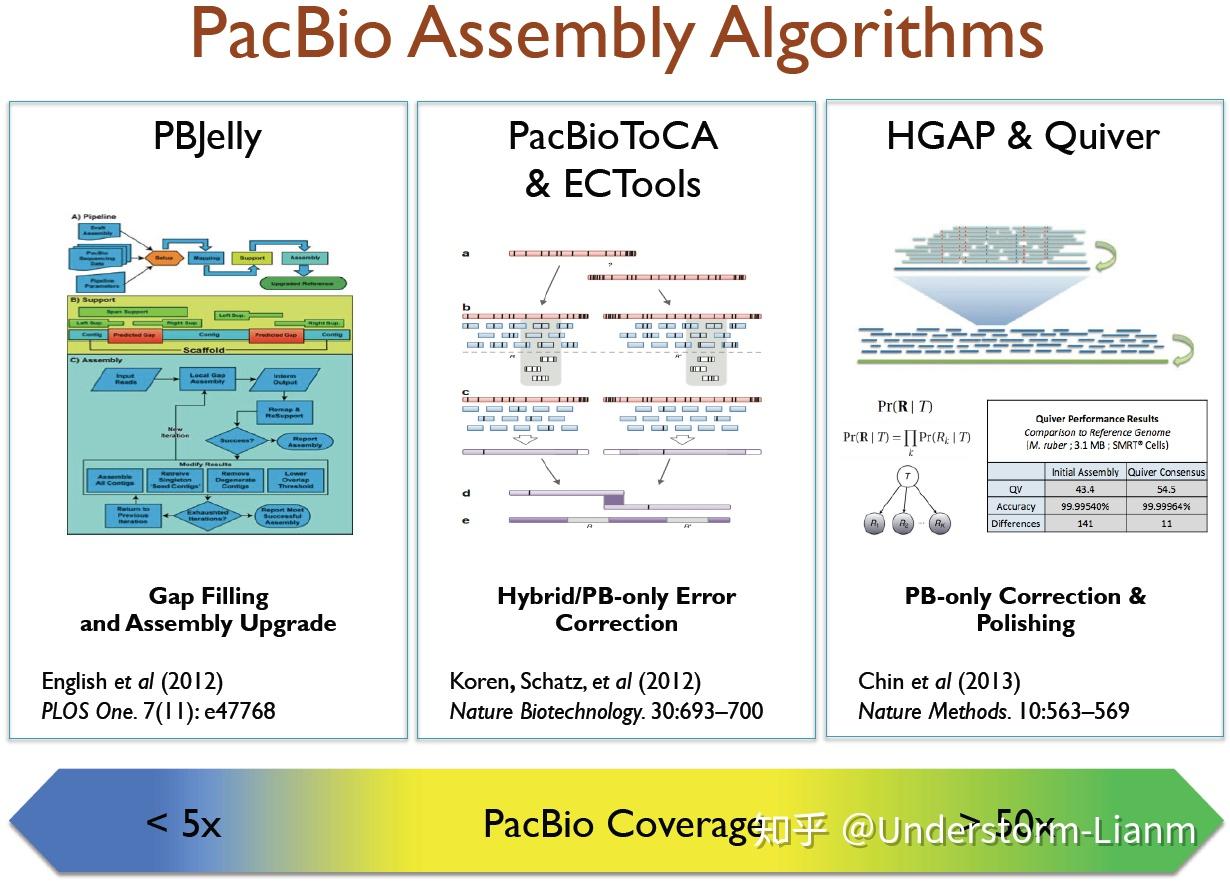
不同的组装策略可以选用的工具:
(1) PacBio-only
HGAP:先进行reads的预组装(preassembly),然后用Celera® Assembler进行进一步组装,最后用 Quiver 进行校正
Falcon:一个试验性的二倍体组装工具,已经在Gb级别大小的基因组上做了试验
Canu:以Celera Assembler为基础,为三代单分子测序而开发出的分支工具
Celera® Assembler:现在,Celera® Assembler 8.1 已经可以直接用于subreads的组装
(2) Hybrid
pacBioToCA:Celera® Assembler的一个error correction模块,最初是用来align short reads to PacBio reads 和 generate consensus sequences。随后,这些错误校正过的PacBio reads可以用Celera® Assembler进行组装
ECTools:使用 unitigs (High quality contigs formed from unambiguous, unique overlaps of reads) 而非short reads进行校正
SPAdes :SPAdes原本是进行短序列组装,在3.0版本后增加了对PacBio的混合组装的支持
Cerulean :用ABySS构建de Bruijn graph,在图的bubbles位置利用PacBio的long reads解决bubbles带来的分支选择问题,从而延伸contigs
(3) Gap Filling
PBJelly 2 :对已经组装过的基因组,用PacBio的long reads进行补洞
2.1. de novo assembly 算法
基因组的组装问题,实际上就是从序列得到的图中搜寻遍历路径的问题,有两种构建图的方法:
- overlap-layout-consensus (OLC)
- de Bruijn graph

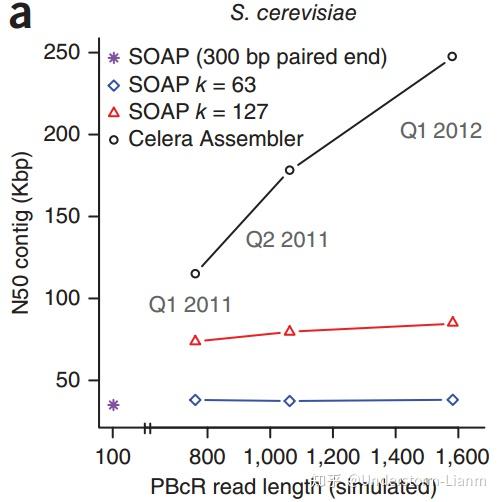
可以看到,随着reads长度的增加,基于OLC算法的组装工具组装出的contigs的长度几乎在线性增长,而基于de Bruijn图算法的组装效果并没有随着reads长度的增
加而提高
3. 测序错误校正
三代单分子测序会产生较高的随机错误,平均正确率在82.1%-84.6%。这么高的错误率显然不能直接用于后续的分析,需要进行错误校正:
- 多测几个pass:由于测序序列是发夹结构,可以进行多轮的滚环测序,靠覆盖度来自我纠错,如果通量不是限制因素,那么PacBio是目前最准确的测序方式:错误率可以无限接近罕见突变的发生率(即无法分辨是测序错误还是罕见突变),不过这会极大缩短有效测序的插入序列的长度
- 用二代的短reads校正:2012年冷泉港实验室的Michael Schatz开发了一种纠错算法,用二代测序的短读长高精确数据对三代长读长数据进行纠错,这种称为”混合纠错拼接”(PBcR (PacBio corrected Reads) algorithm)
(1) Map short reads to long reads
(2) Trim long reads at coverage gaps
(3) Compute consensus for each long read

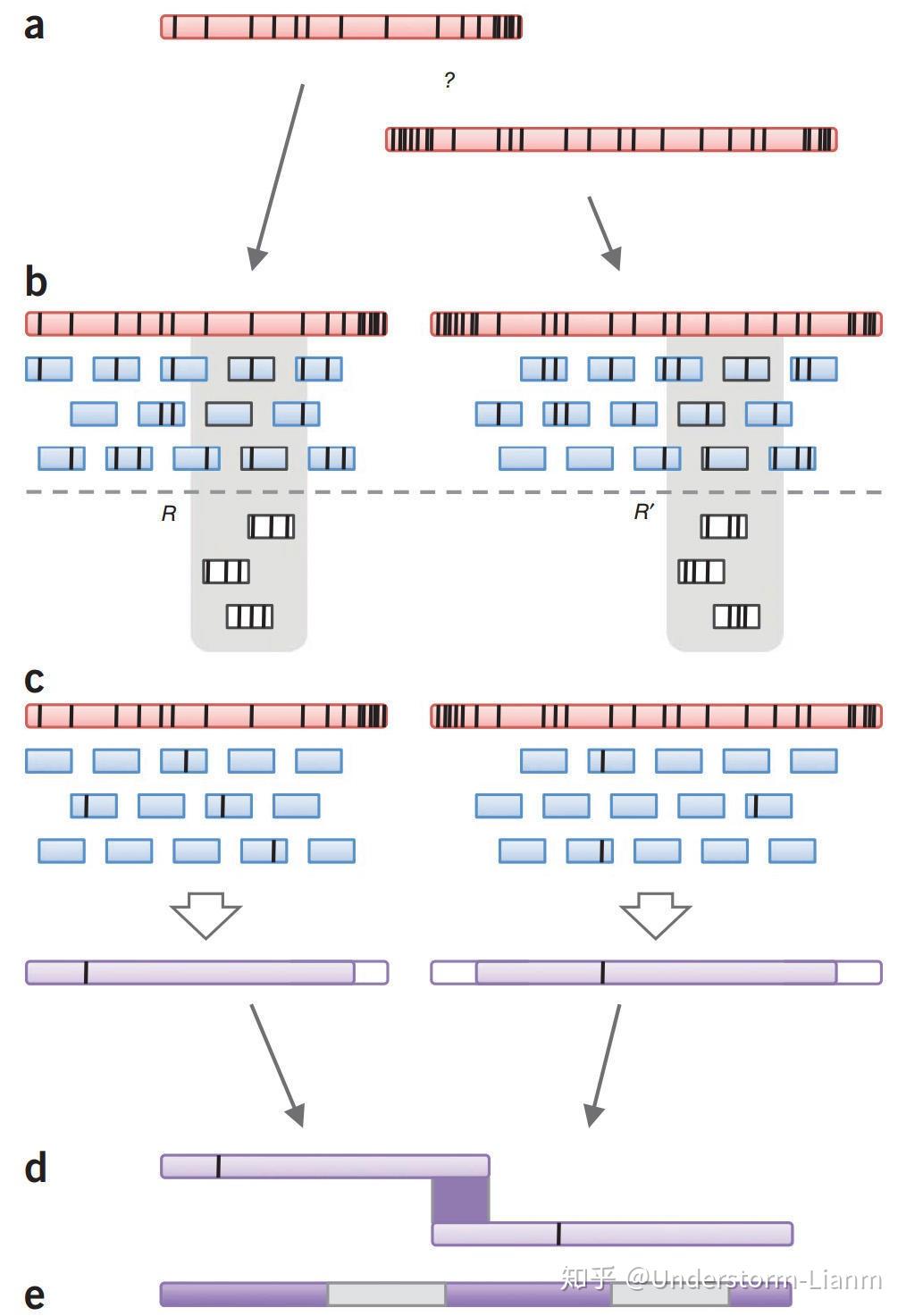
粉色长方形:单个PacBio RS reads;黑色竖线:测序错误;
(a) 由于测序错误碱基的存在使得两条reads就难确定是否在末端重叠;
(b) 高质量的短reads比对到存在错误的长reads;短reads中的黑色竖线表示 ‘mapping errors’ ,是长reads和短reads中测序错误的组合,此外双拷贝的重复序列的存在(灰色轮廓)导致在每一个拷贝中出现短reads的堆挤,为避免reads map到错误的重复区,仅保留最高比对值的短reads;
(c) 剩余的比对形成一致性序列(紫色长方形),长reads和短reads中共有的部分错误未能得到纠正;
(d) overlap纠正后的长reads;
(e) 最后的组装能够跨越重复区域。
校正过程中会将short reads未覆盖到的Gap进行裁剪,short reads在PacBio long reads上的覆盖情况:
这样做的其中一个考虑是去除adapter


那么是什么原因导致了低覆盖度区域的产生的呢?
(1) Simple Repeats – Kmer Frequency Too High to Seed Overlaps
(2) GC Rich Regions – Known Illumina Bias
(3) Error Dense Regions – Difficult to compute overlaps with many errors




为了克服第三中情况导致的高测序错误率区域的低覆盖度,研究人员提出了用Unitigs进行校正的方法


参考资料:
(1) 三代测序--QC篇
(2) PacBio Training: Large Genome Assembly with PacBio Long Reads
(3) Koren S, Schatz M C, Walenz B P, et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads[J]. Nature Biotechnology, 2012, 30(7):693-700.
