Rosetta Centroid 打分函数简介

作者: 吴炜坤
参考1: function-Improved Recognition of Native-Like Protein Structures Using a Combination of Sequence-Dependent and Sequence-Independent Features of Proteins(Centorid Score)
参考2: Assembly of Protein Tertiary Structures from Fragments with Similar Local Sequences using Simulated Annealing and Bayesian Scoring Functions
参考3: Protein Structure Prediction Using Rosetta
参考4: 更多详见: https://www.rosettacommons.org/docs/latest/rosetta_basics/scoring/centroid-score-terms#score-terms_smooth-terms
一、前言
在过去的10年中,Rosetta在许多建模方面获得了突破性的进展,其成功背后的重要核心就是其打分函数系统。无论是在采样、筛选等阶段都程序需要调用它来对一个蛋白的构象进行评估。目前,Rosetta中的打分函数已经发展了近20年,本文将简要概述centroid函数。
二、Centroid模型与历史发展简介
2.1 Centroid与Full-Atom模型
Rosetta的采样策略分为两种阶段: 低分辨率以及高分辨率阶段。在低分辨率中,蛋白质的侧链被Cβ原子和一个粗粒化球所替代,是简化的模型(centroid)。而在高分辨率中氨基酸侧链以全原子的形式展现(fullatom)。


2.2 Centroid的发展历史
为了模拟蛋白质的折叠过程,如果考虑全原子模型往往会造成能量面过于崎岖,导致绝大部分的采样尝试都无法通过Metropolis准则,因此Rosetta在低分辨率阶段使用了粗粒化模型,极大地增加采样效率。
Centroid打分函数的基本框架,最早在1997年由Simons提出,根据贝叶斯定理将蛋白的总能分离成若干项进行计算,最初只考虑蛋白整体的回旋半径(P~Rg~)、氨基酸相互作用对频率分布(P~pairs~)以及不同疏水环境中氨基酸出现的频率(P~env~)三项组成。但是这个模型并无法很好地描述β-sheet结构的细节。
随后在1999年,Simons继续改进先前的模型,提出使用二级结构堆积(packing)替代先前用Rg值来描述蛋白的整体结构, 并引入了描述β-strand配对项(P~sheet~)来描述二级结构排列与天然的类似程度,除此以外还引入了范德华力排斥项(VdW)、cbeta项、Rg项、cenpack项使得Fragment组装的蛋白结构中原子分布接近真实蛋白,避免过于松散或过于紧密。
2004年,Shmygelska引入co、rama、rsigma、hbsrbb以及hblrbb项来继续提升Centroid打分函数在从头预测蛋白质结构的应用效果,其中rama,hbsrbb以及hblrbb均源于Rosetta全原子力场。
以上Centroid框架都是基于骨架参数进行打分,直到Centroid_rot模型的提出,才真正考虑了氨基酸侧链排布问题。
在Centroid基本框架下,经过多年的发展,Centroid已经可应用于Docking、RNA、膜蛋白预测和设计、同源建模优化、Loop建模等领域。
- RNA centroid score terms: RNA相关项
- Membrane centroid score terms: 膜蛋白相关项
- "Interchain" terms: 用于低分辨率的对接时的打分函数
- "Smooth" terms: 在优化x-ray、冷冻电镜结构以及同源建模时使用较多
- Cen_rot terms: 在Cen阶段添加rotamer项,更好地描述侧链排布
三、Centroid的基本框架
用于低分辨率阶段的打分函数(centroid)的基本项(除vdw)都是基于贝叶斯定理统计得到的。
贝叶斯定理:
\\P(A | B)=\frac{P(B | A) P(A)}{P(B)}
P(A|B):在B发生的条件下,A发生的概率
P(B|A):在A发生的条件下,B发生的概率
P(B): B发生的概率
P(A): A发生的概率
于是我们可以将蛋白质结构评分的问题转化成求解序列与结构之间的概率问题。
\\\begin{array}{l}{P(\text { structure } | \text { sequence })=P(\text {structure}) \times \frac{P(\text {sequence} | \text {structure})}{P(\text {sequence})}}\end{array}
用通俗语言阐述这个公式就是求解: 当给定一条序列时(sequence),我们用Rosetta通过Fragment片段拼接出来的结构(structure)就是该序列天然结构的概率:P(structure|sequence)。但是这个概率我们并没有办法直接求解,那么就需要对这个问题进行转化,用我们容易从数据库中获取的概率来计算它。比如,通过贝叶斯定理转化后,我们就只需要求解:
- 在给定一个模拟结构的前提下,该序列折叠后就是这个结构的概率P(sequence|structure)
- 该结构的结构的出现概率P(sturcture)
- 序列在天然中出现的概率P(sequence)。因为在比较不同结构的打分时,序列都是一致的。因此P(sequence)作为常量处理。
1 P(sequence|structure)部分:
求解这个概率的本质就是在计算给定氨基酸序列与给定结构之间的适配性,而这个指标与蛋白质结构能量是直接相关的,如果给定的序列与结构骨架的适配性高,那么那么序列中氨基酸之间的相互作用质量越高。
通常在求解P(sequence|structure)时会做两种近似假设:
第一种假设: 氨基酸序列中每一个位点的氨基酸都是独立的,相互不影响,氨基酸类型只和局部结构E有关(大多以二级结构或所处的疏水环境来衡量),但是这种假设忽略了相互作用,但是能很好地描述局部的疏水环境。
\\P(\text { sequence } | \text { structure }) \cong \prod_{i} P\left(a a_{i} | E_{i}\right)
第二种假设: 前提基础是认为氨基酸两两之间的相互作用足以描述折叠自由能,并且认为氨基酸序列中每两对位点的氨基酸都是独立的,氨基酸类型只与氨基酸之间的距离有关。这种假设可以很好"隐式地"近似了分子内部的相互作用,如静电相互作用,氢键相互作用,溶解自由能等等。但是缺点是对势的统计分布与数据中蛋白质的大小有直接关系。
\\P(\text { sequence } | \text { structure }) \cong \prod_{i<j} P\left(a a_{i}, a a_{j} | r_{i j}\right)
由于两种假设模型各有优劣,因此在Rosetta中,P(sequence|structure)是使用了联合概率的扩展式进行二阶近似处理为P(env)和P(pairs)两项的乘积。这样近似有个好处,就是将环境和对势较好地结合起来,可以对氨基酸对势进行校正,分别统计处于蛋白质内核和外部环境中氨基酸的相互作用。
\\\begin{array}{l}{P\left(a a_{1}, a a_{2}, \ldots, a a_{n} | \text { structure } \cong \prod_{i} P\left(a a_{i} | E_{i}\right)\right.} {\quad \times \prod_{i<j} \frac{P\left(a a_{i}, a a_{j} | r_{i j}, E_{i}, E_{j}\right)}{P\left(a a_{i} | r_{i j}, E_{i}, E_{j}\right) P\left(a a_{j} | r_{i j}, E_{i}, E_{j}\right)}}\end{array}
\\P(\text { sequence } | \text { structure }) \approx P_{\text { env }} P_{\text { pair }}
env项: 在不同疏水环境中氨基酸出现的概率,假设认为一个氨基酸周围的氨基酸数目越多,那么这个环境的疏水性就越强,因此通过统计一定范围内邻居氨基酸的数据来近似环境的效应。其中nb的统计区间分为: 1, 2, 3, …30, 30+。用于近似描述每个氨基酸类型的溶解自由能。
\\env = \sum_{i}-\ln \left[P\left(\mathbf{aa}{i} | \mathbf{n b}{i}\right)\right]
pairs项: 统计在不同氨基酸的centroid–centroid原子(一级序列需要间隔8个氨基酸以上)的距离区间内,特定氨基酸出现的概率。其中d的统计范围为: 10–12, 7.5–10, 5–7.5, <5埃,这四个区间。用于描述静电相互作用和二硫键连接。
\\pairs = \sum_{i} \sum_{j>i}-\ln \left[\frac{P\left(\mathrm{aa}{i}|s{i j} d_{i j}\right)}{P\left(\mathrm{aa}{i} | s{i j} d_{i j}\right) P\left(\mathrm{aa}{i} | s{i j} d_{i j}\right)}\right]
由此我们可以顺利地计算出P(sequence|structure)最相关的两项。
2 P(structure)部分:
这部分的计算前提是基于蛋白质折叠的漏斗模型,认为天然蛋白在折叠过程中总是倾向于能量最低的状态,因此能量越低的结构,它出现概率就越高。换句话说,我们需要求解的P(structure)是给定的结构在折叠过程中出现的概率。如果它和已知天然的结构特征越相似,那么它出现的概率自然也越高。
计算上通常使用一个可以区分native与non-native构象差异的特征作为统计量。比如在第一代Centroid打分函数中,使用的是蛋白质回旋半径值(Rg)作为判断指标,因为通过统计发现,native的rg值通常较小,如果rg值越大,那么这个蛋白是天然结构的概率也就越低。
在第二代Centroid框架下采用多个二级结构向量模型来代表一个结构整体特征,如二级结构之间朝向、距离、β-strand之间配对程度等等位指标。
2.1 向量模型的定义与坐标系
在第二代的Centroid框架下,二级结构向量模型由任意连续的两个氨基酸(二联体、dimmer)作为单位来定义。
比如: 现在有一段α-螺旋和β-strand,如果需要计算向量之间的信息,我们就需要将各个二级结构划穷举分为若干个dimmer,并计算由dimmer定义的向量。在实际中,strand和helix的向量定义有些不同:
- 在β-strand中以两个氨基酸长度作为一个向量生成单位,向量由第一个氨基酸的N原子至第二个氨基酸的羧基氧原子。
- 在一段α-helix中,以每4个氨基酸长度作为一个向量生成单位,前11个和后11个骨架原子坐标的平均值作为向量的起点和终点,以确保单位向量经过局部螺旋片段的中心。
为了正确描述两个二级结构之间的朝向问题,还需要根据v1和v2向量来定义坐标系,可以得到5个变量来描述它们之间的关系(如下图)

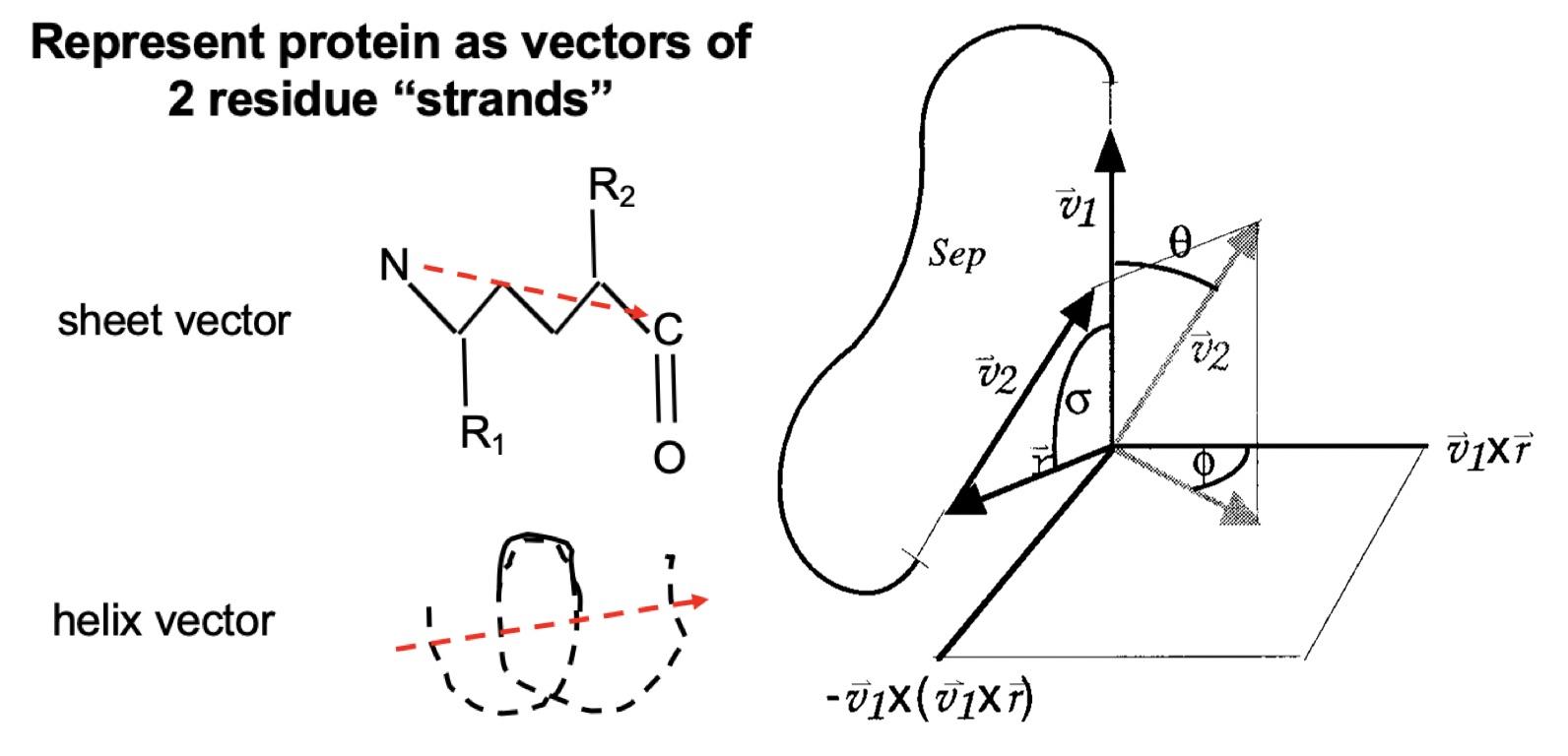
- r: 以两个二级结构向量中点为起始和终止的向量(N-C顺序)
- σ: 为r、v1向量之间的角度
- θ: v2向量在yz坐标平面的投影与z轴的夹角(每36°作为统计区间)
- Φ: v2向量在xy坐标平面的投影与x轴的夹角(每36°作为统计区间)
- Sep: 两个相连的二级结构单元之间Loop的氨基酸长度,用这个度量比用dimmer之间的氨基酸数量描述更清晰一些。
- hb: 以r向量与第一个二级结构片段的C=O(羧基碳氧键)向量的点乘以及r向量与第二个二级结构片段的O=C向量点乘之和来描述(β折叠片之间的氢键),加和后值越接近2,说明两个β-折叠片之间的越处于同一个平面。
开发人员Simons通过统计发现,θΦ、σ、hb之间的分布是相互独立的。因此该联合条件概率分布的近似展开可得:
\\\begin{array}{r}{P_{x x}(r, \phi, \theta, \sigma, h b | S e p) \approx P_{x x-\phi \theta}(\phi, \theta | r, S e p) P_{x x-h b}(h b | r, S e p)} \ {P_{x x-\sigma}(\sigma | r, S e p) P_{x x-d i s t}(r | S e p)}\end{array}
- 其中xx=HH, HS, 或SS,代表helix-helix、helix-strand、strand-strand之间的堆积。
通过测试表明,helix-helix堆积项无法很好地区分native和non-native结构的差异,因此被省略。Centroid打分函数仅保留下helix-strand(HS项)、strand-strand(SS项)之间的堆积以及sheet项来描述P(structure)。
2.2 ss_pair项
通过对strand-strand的详细统计,开发者发现P(hb|r, Sep)以及P(Φ,θ|Sep,r)更有价值,而P(σ|Sep,r)项的分布无法很好的区分native、non-native结构。因此产生两种计算策略:
SchemeA: ss_pair = SSθΦ+SShb+SSd
SchemeB: ss_pair = SSθΦ+SShb+SSdσ
P(Φ,θ|r, Sep, s)的计算:
\\\mathrm{SS}{\phi, \theta}=\sum{m} \sum_{n>m}-\ln \left[P\left(\phi_{m n}, \theta_{m n} | d_{m n}, \mathrm{sp}{m n}, s{m n}\right)\right]
P(r|s)的计算:
\\\mathrm{SS}{d}=\sum{m} \sum_{n>m}-\ln \left[P\left(d_{m n} | s_{m n}\right)\right]
P(hb|r, s)的计算:
\\ \mathrm{SS}{h b}=\sum{m} \sum_{n>m}-\ln \left[P\left(\mathrm{hb}{m n} | d{m n}, s_{m n}\right)\right]
P(r,σ|ρ)的计算:
\\\mathrm{SS}{d \sigma}=\sum{m} \sum_{n>m}-\ln \left[\mathbf{P}\left(d_{m n},\sigma_{m n} | \rho_{m}, \rho_{n}\right)\right]
其中:
- m和n是两个dimmer的序号
- sp为两段strand上dimmer之间的氨基酸间隔, 以小于2、2–10、大于10个氨基酸长度作为统计区间
- s为两个dimmer之间的氨基酸间隔长度, 以大于5或大于10个氨基酸长度作为统计区间
- d为上述的r向量的距离,其长度应小于6.5埃
- ρ代表r向量与C=O向量、以及r向量与O=C向量角度的平均值,以180°作为一个统计区间。
- σ角以18°作为一个统计区间
2.2 hs_pairs项
该项描述β-strand与螺旋之间的堆积。在该项中,由于螺旋结构本身的圆柱对称性,与z轴旋转相关的项均忽略不计。
\\hs_pairs = \sum_{m} \sum_{n}-\ln \left[P\left(\phi_{m n}, \theta_{m n} | s p_{m n} d_{m n}\right)\right]
- d为上述的r向量的距离,其长度应小于12埃
- sp为helix和strand上dimmer的氨基酸序列间隔,统计区间为 小于2, 2–10, 大于10 个氨基酸长度
2.3 sheet项
为了更好地表述β折叠配对的情况,引入了sheet相关的统计项,在已知结构中β-strand数量的前提下,统计β-strand配对成sheet数量的概率。
\\\begin{array}{l}{P_{\text { sheet }}(\text { sheet configuration } | \text {number of strands) }} {=\exp \left(c_{n}+a_{1} n_{1}+a_{2}n_{2}\right)}\end{array}
\\sheet = -\ln \left[P\left(n_{\text { sheets }},n_{\text { lonestrands }} | n_{\text { strands }}\right)\right]
其中,
- c~n~是标准化常数,依据strand数量不同而变化,c~n~ = 0.07, 0.41, 0.43, 0.60, 0.61, 0.85, 0.86, 1.12
- n~1~是结构中β-strand的总数
- n~2~是结构中β-strand为配对的总数
- a~1~=-0.9/a~2~=-2.7
2.4 VdW项
为了避免原子间过度堆积导致的化学立体构象冲撞,引入了范德华的排斥项。根据不同原子类型,计算结构中每对原子距离:
\\VdW = \sum_{i} \sum_{j>i} \frac{\left(r_{i j}^{2}-d_{i j}^{2}\right)^{2}}{r_{i j}} ; d_{i j}<r_{i j}
- d为原子间的距离
- r为两个原子的范德华半径之和
3 附加校正项
除了贝叶斯推导衍生的打分项,Rosetta中还添加了额外的附加项。
3.1 rg、cbeta与cenpack项
rg(Radius of gyration)、cbeta和cenpack项都用于描述一个蛋白折叠的紧密度。
rg项: 通过计算结构中每对氨基酸之间的距离均方根来描述:
\\rg = \sqrt{\left\langle d_{i j}^{2}\right\rangle}
cbeta项: 通过统计每个Cβ原子与周围Cβ的数量,确定nb值并带入公式, 计算整体结构Cβ原子的密度分布均值。
\\cbeta = \sum_{i} \sum_{s h}-\ln \left[\frac{P_{\text { compact }}\left(\mathrm{n} \mathrm{b}{i, s h}\right)}{P{\text { random }}\left(\mathrm{n} \mathrm{b}_{i, s h}\right)}\right]
- nb: 代表每一个残基10埃范围内的Cβ原子数量。
- P~compact~(nb~i,sh~): 代表在从Fragment组装而来且具有紧密结构中,统计每一个氨基酸在shell范围内的Cβ原子数量的概率分布。
- P~random~(nb~i,sh~): 代表在从Fragment随机组装而来的结构中,统计每一个氨基酸在shell范围内的Cβ原子数量的概率分布。
- 通常shell范围半径选为: 6和12埃区间进行统计。
cenpack项:
通过统计整个蛋白所有的centroid-centroid原子对类型距离的分布情况,与PDB统计和decoy训练的分布情况作比较,类似cbeta。(此项缺乏详细的信息)
3.2 rsigma
根据strand dimmer之间的距离进行的统计,其中包括考虑register部分。(此项缺乏更详细的信息)


3.3 co
Contact Order是计算蛋白质内所有氨基酸相互作用对在一级序列上的平均间隔距离除以总氨基酸长度的指标。
\\CO=\frac{1}{L \cdot N} \sum^{N} \Delta S_{i, j}
- N 为氨基酸相互作用对的总数
- ΔSi,j 为两个氨基酸相互作用对,i和j氨基酸在一级序列上的间隔距离
- L 为氨基酸的总长度
通常在一个单结构域的蛋白中,CO分布在5%-25%。螺旋蛋白的CO较低,而β折叠片为主的蛋白CO较高。
3.4 rama
与骨架二面角(phi,psi)分布统计相关的项。
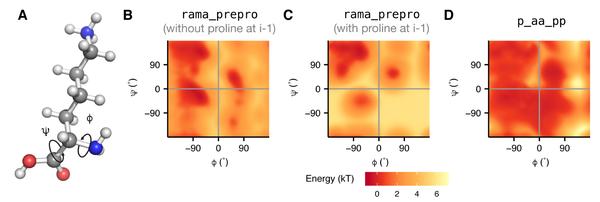

直接从数据库中得到的统计量,其含义为给定一种氨基酸类型时,其骨架二面角的概率分布,分为i+1为脯氨酸和非脯氨酸两种情况。
如果模型中二面角落于频率分布高的格点区域,则能量越低,骨架构象合理的可能性越高。当偏离这些高频率分布的区域时,骨架二面角的能量越高。
\\E_{\text { rama pre-pro }}=\Sigma_{i}\left\{\begin{array}{cc}{-\ln \left[P_{\text { reg }}\left(\phi_{i}, \psi_{i} | \text { aa }_{i}\right)\right]} \\ {-\ln \left[P_{\text { prepro }}\left(\phi_{i}, \psi_{i} | \text { aa }_{i}\right)\right]}\end{array}\right.
- P(reg)为C端或i+1为非脯氨酸的概率
- P(prepro)为i+1为脯氨酸时的概率
3.5 hb_srbb/hb_lrbb
hbond项是根据Top8000高精度结构数据库中衍生得到的,首先通过将蛋白质内部的极性相互作用对的分离,并拟合了它们的二面角、键角、供体受体原子的距离等参数的能量分布。
目前hbond项与4个自由度有关: BAH、 B2BAH、 HA、 AHD,根据原子轨道的杂化情况不同, BAH与 B2BAH拟合成两种类型的能量密度分布,受体原子杂化类型由 设置。
E_{\mathrm{hbond}}=\sum_{H, A} w_{H} w_{A} f\left(E_{\mathrm{hbond}}^{H A}\left(d_{H \\A}\right)+E_{\mathrm{hbond}}^{A H D}\left(\theta_{A H D}\right)+E_{\mathrm{hbond}}^{B A H}\left(\theta_{B A H}\right)+E_{\mathrm{hbond}}^{B_{2} B A H}\left(\rho, \phi_{B_{2} B A H}, \theta_{B A H}\right)\right)
氢键项的能量同时与供体和受体的原子类型有关,不同原子的权重由ωA和ωH设置。

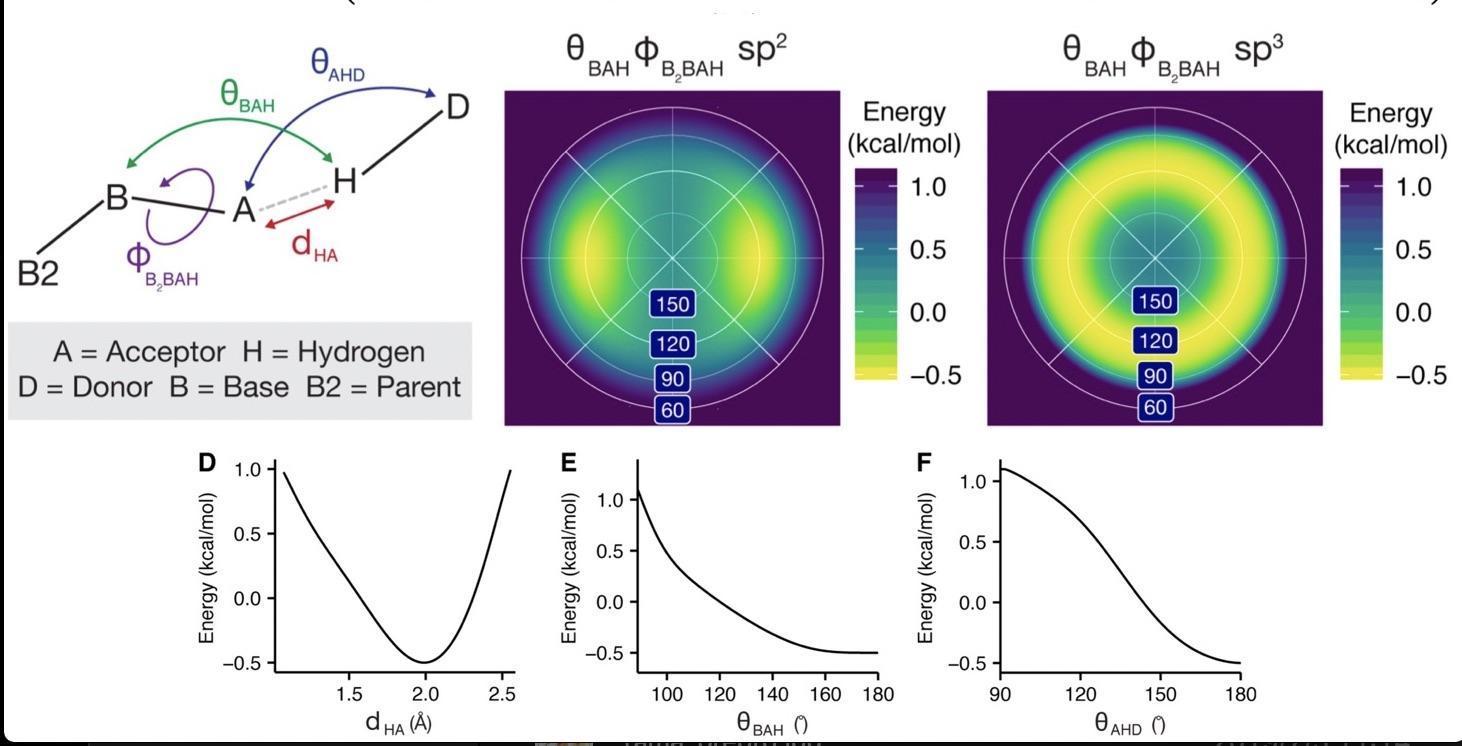
最后通过函数f(x)进行平滑处理, 避免非连续性:
\\f(x)=\left\{\begin{array}{cc}{x} & {x<-0.1} \\ {-0.025+\frac{x}{2}+2.5 x^{2}} & {-0.1 \leq x<0.1} \\ {0} & {0.1 \leq x}\end{array}\right.
具体的hbond项类型可分为:
- hbond_sr_bb: 短距离骨架氢键,如α螺旋的骨架氢键项
- hbond_lr_bb: 远距离的骨架氢键,如β折叠或loop中的骨架氢键项
- hbond_sc_bb: 侧链与骨架之间的氢键项
- hbond_sc: 侧链与侧链之间的氢键项
为了避免重复计算,如果骨架与骨架之间已经产生了氢键,那么侧链与骨架原子之间的氢键计算被排除。
5 总分计算
Centroid的总能(Total energy)是各个项组成的加权线性拟合得到的结果。
# 总能的计算
Energy = w1*term1 + w2*term2 + w3*term 3 + …