
RNA-seq:转录组数据分析处理(上)

by:superqun
一、流程概括
- RNA-seq的原始数据(raw data)的质量评估
- linux环境和R语言环境
- raw data的过滤和清除不可信数据(clean reads)
- reads回帖基因组和转录组(alignment)
- 计数(count )
- 基因差异分析(Gene DE)
- 数据的下游分析
二、准备工作
- 学习illumina公司测序原理
- 测序得到的fastq文件
- 注释文件和基因组文件的准备
1. fastq测序文件
在illumina的测序文件中,采用双端测序(paired-end),一个样本得到的是seq_1.fastq.gz和seq_2.fastq.gz两个文件,每个文件存放一段测序文件。在illumina的测序的cDNA短链被修饰为以下形式(图源见水印):


两端的序列是保护碱基(terminal sequence)、接头序列(adapter)、索引序列(index)、引物结合位点(Primer Binding Site):其中 adapter是和flowcell上的接头互补配对结合的;index是一段特异序列,加入index是为了提高illumina测序仪的使用率,因为同一个泳道可能会测序多个样品,样品间的区分就是通过index区分。参考:illumina 双端测序(pair end)、双端测序中read1和read2的关系。
在illumina公司测得的序列文件经过处理以fastq文件协议存储为*.fastq格式文件。在fastq文件中每4行存储一个read。
第一行:以@开头接ReadID和其他信息,分别介绍了 第二行:read测序信息 第三行:规定必须以“+”开头,后面跟着可选的ID标识符和可选的描述内容,如果“+”后面有内容,该内容必须与第一行“@”后的内容相同 第四行:每个碱基的质量得分。记分方法是利用ERROR P经过对数和运算分为40个级别分别与ASCII码的第33号!和第73号I对应。用ASCII码表示碱基质量是为了减少文件空间占据和防止移码导致的数据损失。fastq文件预览如下:


2.注释文件和基因组文件的获取
- 基因组获取方式:可以从NCBI、NCSC、Ensembl网站或者检索关键词“hg38 ftp UCSC” 人类基因组hg38.fa.gz大概是938MB左右。文件获取可以点击网站下载。
可以通过云盘的离线下载来加速下载进程 - 基因组的选择:以Ensembl网站提供的基因组为例,比对用基因组应该选择
Homo_sapiens.GRCh38.dna.primary_assembly.fa - Ensembl基因组的不同版本详见README和高通量测序数据处理学习记录(零):NGS分析如何选择合适的参考基因组和注释文件
三、软件安装
- 安装方式:软件安装可以通过例如
apt-get、miniconda等方式来安装。由于miniconda的便捷行,使用conda进行如下软件的安装。 - 软件列举 质控:
fastqc ,multiqc , trimmomatic, cutadapt, trim-galore比对:star , hisat2 , tophat , bowtie2 , bwa , subread计数:htseq , bedtools, salmon - miniconda的安装:
- 可以通过点击清华大学开源软件站或者检索“清华大学 conda”访问镜像网站(清华镜像站因为服务器在中国访问速度比较快),点击Anoconda界面,选择Miniconda下载安装,windows在安装好需要设置环境变量。
- linux测试Miniconda的安装:
conda -v - 创建名为rna的环境变量:
conda -n rna python=2(许多软件依赖python2环境) - 配置conda,添加镜像源头:输入如下命令
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --set show_channel_urls yes
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/- 软件安装:
conda install <software>会自动安装软件和软件环境。值得注意的是需要在rna的环境变量下安装以上软件。激活rna环境变量的代码:
source activate rna四、质量汇报生成与读取
1.fastq质量汇报
使用命令fastqc -o <output dir> <seqfile1,seqfile2..>来进行质量报告。每个fastqc文件会获得一个质量分析报告,来描述此次RNA-seq的测序质量。 获取质量报告如图:


Basic Statistics
从read水平来总览,判断测序质量。 Encoding :测序平台的版本,因为不同版本的 error p的计算方法不一样。 Total sequence:测序深度。一共测序的read数。是质量分析的主要参数。 Sequence length:测序长度。 %GC:GC碱基含量比,一般是物种特异性,比如人类是42%左右。
Perbase sequence quality
横坐标: 第1-100个测序得到的碱基 纵坐标: 测序质量评估。这里的Q=-10*lg10(error P),即20%代表1%的错误读取率,30%代表0.1%的错误读取率 箱型图: 红色线,是某个顺序下测序碱基所有测序质量的中位数。黄色块,是测序质量在25%-75%区域。蓝色线,平均数。 一般要求: 测序箱型图10%的线大于Q=20。Q20过滤法。
per tail sequence quality
横坐标:同上。 纵坐标:tail的index编号。 目的:防止测序过程中某些tail受不可控因素测序质量低。 标准:蓝色表示质量高,浅色或暖色表示质量低,后续的分析可以去除低质量tail。


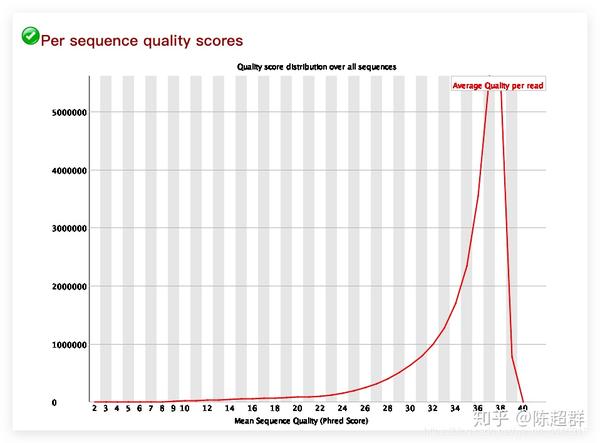
Per sequence quality scores
从read的总体测序质量分布来判定此次的测序质量,是质量分析的重要标准之一。 横坐标:表示read的测序质量Q=-10*lg10(error P)。 纵坐标:表示在该Q值下的read 的数量 标准:需要集中在高分区

Per base sequence content
横坐标:1-100的测序碱基位置 纵坐标:碱基百分比 标准:理论上,ATCG碱基的分布应该差别不大,即四条线应该大致平行状态。如果AT或CG差异超过10%,此项检测是危险的。一般是测序机器前几个碱基测序时候因为状态调整导致测序略有偏差,如果前几个碱基偏差较大,可以在后期将前几个碱基切掉。


Sequence Length Distribution
统计read的碱基长度,本例理论上测序应该全是100bp。 横坐标:是read的碱基长度 纵坐标:是该长度下的read数量


Per sequence GC content
横坐标:每个read的平局GC含量占比 纵坐标:一定GC比下的read数 标准:蓝色是理论值,红色是真实值。两者接近是比较好的状态。如果有双峰,可能混有了其他物种的DNA序列。


Adapter Content
一般测序在初步生成fastq文件时候,adapter会被去除,但是有的会没有去除或者遗漏部分adapter。所以这一步是检测RNA-seq测序过程中adapter是否去除。如果没有去除会严重影响后续的比对工作。没有去除的adapter在质量处理环节会被处理掉。


2. multiqc质量报告
multiqc可以对几个fastqc报告文件进行总结并汇总到一个报告文件中,以更直观到防止展示。使用方法
multiqc <analysis directory>

五、数据处理
数据处理内容:fiter the bad quality reads and remove adaptors. 处理软件:数据到处理可以使用多款软件,trim_galore在各文献中表现良好。
1.trim_galore 的使用方法
trim_galore:可以处理illumina,nextera3,smallRNA测序平台的双端和单端数据,包括去除adapter和低质量reads。 trim_galore的参数: trim_galore的参数在处理过程比较重要:
trim_galore [options] <filename>
--quality<int> #设定phred quality阈值。默认20(99%的read质量),如果测序深度较深,可以设定25
--phred33 #设定记分方式,代表Q+33=ASCII码的方式来记分方式。这是默认值。
--paired # 对于双端结果,一对reads中若一个read因为质量或其他原因被抛弃,则对应的另一个read也抛弃。
--output_dir #输出目录,需确保路径存在并可以访问
--length #设定长度阈值,小于此长度会被抛弃。这里测序长度是100我设定来75,感觉有点浪费
--strency #设定可以忍受的前后adapter重叠的碱基数,默认是1。不是很明白这个参数的意义
-e<ERROR rate> #设定默认质量控制数,默认是0.1,即ERROR rate大于10%的read 会被舍弃,如果添加来--paired参数则会舍弃一对reads
<filename> #如果是采用illumina双端测序的测序文件,应该同时输入两个文件。构建命令:
trim_galore -output_dir clean --paired --length 75 --quality 25 --stringency 5 seq_1.fasq.gz seq_2.fastq.gz处理需要花上一定时间和磁盘空间。得到处理后数据


2. 整理后数据的质量分析。
对过滤后对文件进行质量分析。观察过滤结果。同样使用fastqc和multiqc两个软件进行质量分析。得到结果如下:

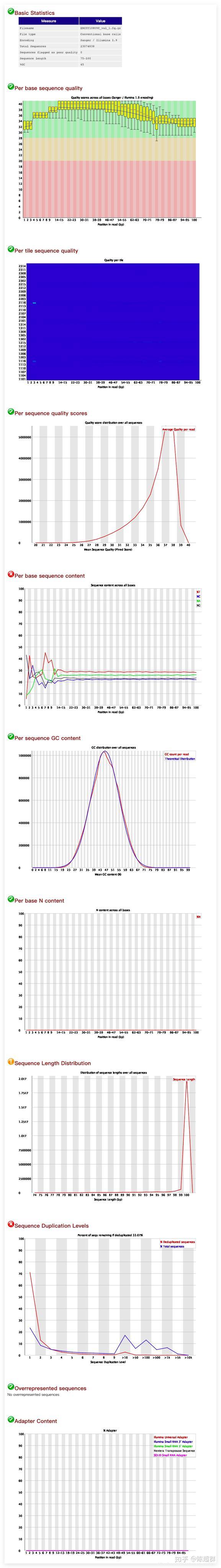
ENCFF108UVC_val_1_fastqc的质量报告
观察到总read数减小和总体read的质量变高,小部分adapter也被去除。更具体过滤和trim_galore的数据处理情况可以在seq_trimming_report.txt中查看。
SUMMARISING RUN PARAMETERS
==========================
Input filename: ENCFF108UVC.fastq.gz
Trimming mode: paired-end
Trim Galore version: 0.5.0
Cutadapt version: 1.18
Quality Phred score cutoff: 25
Quality encoding type selected: ASCII+33
Adapter sequence: 'AGATCGGAAGAGC' (Illumina TruSeq, Sanger iPCR; auto-detected)
Maximum trimming error rate: 0.1 (default)
Minimum required adapter overlap (stringency): 5 bp
Minimum required sequence length for both reads before a sequence pair gets removed: 75 bp
Output file will be GZIP compressed
This is cutadapt 1.18 with Python 2.7.6
Command line parameters: -f fastq -e 0.1 -q 25 -O 5 -a AGATCGGAAGAGC ENCFF108UVC.fastq.gz
Processing reads on 1 core in single-end mode ...
Finished in 1038.93 s (40 us/read; 1.50 M reads/minute).
=== Summary ===
Total reads processed: 26,038,229
Reads with adapters: 714,205 (2.7%)
Reads written (passing filters): 26,038,229 (100.0%)
Total basepairs processed: 2,603,822,900 bp
Quality-trimmed: 82,577,636 bp (3.2%)
Total written (filtered): 2,513,138,030 bp (96.5%)由报告可以知道处理的具体详情。
六、比对回帖
概况:使用处理后的fastq文件和基因组与转录组比对,确定在转录组或者基因组中的关系。在转录组和基因组的比对采取的方案不同。分别是ungapped alignment to transcriptome和Gapped aligenment to genome。 软件:hisat2和STAR在比对回帖上都有比较好的表现。有文献显示,hisat2在纳伪较少但是弃真较多,但是速度比较快。STAR就比对而言综合质量比较好,在长短reads回帖上都有良好发挥。由于hisat2的速度优势,选择hisat2作为本次比对的软件。 在比对之前首先要先进行索引文件的获取或者制作。
1. 索引文件的获取
- 不同的比对软件构建索引方式不同,所用的索引也不尽相同
- 索引文件可以去网站下载也可以自己构建。但是索引构建会比较费时间。建立索引文件需要大约一个小时(MAC: 2.6 GHz Intel Core i5/ 8 GB 1600 MHz DDR3) 。
- 网站下载hisat2基因组索引:http://ccb.jhu.edu/software/hisat2/index.shtml
- 本地索引文件构建参考了CSDN@ Richard_Jolin的构建过程
- 索引文件的格式如下,是由多个文件构成,要保证索引文件的格式和名称部分一致。


2. hisat2的比对回帖
使用hisat2回帖
公式构建根据hisat2 的使用说明书构建了以下公式:
hisat2 -p 6 -x <dir of index of genome> -1 seq_val_1.fq.gz -2 seq_val_2.fq.gz -S tem.hisat2.sam参数说明:
-p #多线程数 -x #参考基因组索引文件目录和前缀 -1 #双端测序中一端测序文件 -2 #同上 -S #输出的sam文件
说明:在比对过程中,hisat会自动将双端测序匹配同一reads并在基因组中比对,最后两个双端测序生成一个sam文件。比对回帖过程需要消耗大量时间和电脑运行速度和硬盘存储空间。5G左右fastq文件比对回帖过程消耗大概一个小时,生成了17G的sam格式文件。回帖完成会生成一个回帖报告。


samtools 软件进行格式转换
SAM文件和BAM文件 samtools 是针对比对回帖的结果——sam和bam格式文件的进一步分析使用的软件。sam格式文件由于体量过大,一般都是使用bam文件来进行存储。由于bam文件是二进制存储所以文件大小比sam格式文件小许多,大约是sam格式体积的1/6 。 samtools将sam转换bam文件
samtools view -S seq.sam -b > seq.bam #文件格式转换
samtools sort seq.bam -0 seq_sorted.bam ##将bam文件排序
samtools index seq_sorted.bam #对排序后对bam文件索引生成bai格式文件,用于快速随机处理。至此一个回帖到基因组对RNA-seq文件构建完成。这个seq_sourted.bam文件可以通过samtools或者IGV( Integrative Genomics Viewer)独立软件进行查看。在IGV软件中载入seq_sourted.bam文件。 可以很直观清晰地观察到reads在基因组中的回帖情况和外显子与内含子的关系。


3.对回帖bam文件进行质量评估。
samtools falgstate :统计bam文件中比对flag信息,然后输出比对结果。 公式:
samtools flagstate seq_sorted.bam > seq_sorted.flagstate结果如下
47335812 + 0 in total (QC-passed reads + QC-failed reads) 3734708 + 0 secondary 0 + 0 supplementary 0 + 0 duplicates 46714923 + 0 mapped (98.69% : N/A) 43601104 + 0 paired in sequencing 21800552 + 0 read1 21800552 + 0 read2 42216752 + 0 properly paired (96.82% : N/A) 42879780 + 0 with itself and mate mapped 100435 + 0 singletons (0.23% : N/A) 337412 + 0 with mate mapped to a different chr 308168 + 0 with mate mapped to a different chr (mapQ>=5)
七、count
计算RNA-seq测序reads对在基因组中对比对深度。 计数工具:feature counts 公式构建:
feature counts -T 6 -t exon -g gene_id -a <gencode.gtf> -o seq_featurecount.txt <seq.bam>参数:
-g # 注释文件中提取对Meta-feature 默认是gene_id
-t # 提取注释文件中的Meta-feature 默认是 exon -p #参数是针对paired-end 数据 -a #输入GTF/GFF 注释文件 -o #输出文件
接下来是表达矩阵构建。在R语言环境下分析。
共勉!欢迎大家踊跃交流,讨论,质疑,批评。留言必回。
后续会更新:利用TCGA数据库进行数据挖掘 ; RNAseq的下游分析 ; 生物信息学的基础知识荟萃...
我想建立并管理一个高质量的生信&统计相关的微信讨论群,如果你想参与讨论,可以添加微信:veryqun 。我会拉你进群,当然有问题也可以微信咨询我。
因为我的笔记分布CSDN、简书、知乎专栏等比较零散,管理起来比较麻烦,思考再三申请了一个微信公众号,会更加方便地发布更多有关生信息、统计方面内容,如果你觉得有需要可以尝试性和我缔结契约 ^ . ^ 。公众号是:进击的大肠杆菌

