
技术分享 | 单细胞RNA测序的实验设计参考

01
背景介绍
目前的转录组测序技术只能获得群体的遗传或表达信息,而无法区分细胞间的差异,单细胞测序技术可以研究单个细胞或基因的表达情况,从而研究细胞异质性,也可以获得基因表达、剪接等信息以及根据这些信息构建的调控关系。因此,在实验之前要进行合理、细致的实验设计,避免一些混杂因素(confounding factors)的影响并基于生物学的变异得出可靠的结论,防止批量操作时引入人工产物影响实验结果。
本文基于两种主流的单细胞RNA测序技术:Smart-seq2和10X Genomics 3’ sequencing,讨论了单细胞转录组测序的实验设计原则和注意事项,并对两种方法的优缺点进行比较分析。
02
两种方法:Smart-seq2和10X Genomics 3’sequencing的比较
(1)SMART技术:Smart技术是基于高保真的反转录酶、模板转换和前置放大来增加cDNA得率,实验流程2天,得到的是全长转录本。该方法有较好的覆盖范围,可检测到稀有转录本,不需要额外的专业设备,因此应用范围较广。但是该方法不适合大批量实验(如96甚至384个样本)。
(2)10X Genomics 3’sequencing :该方法基于微流控技术(Microfluidics-based approaches),与Smart有相似的分子生物学原理,运用了模板转换技术,但与Smart的细胞捕获和通量不同。droplet-based方法是将单个细胞包裹在一个小油滴中(含有barcode和RT primer)反转录成cDNA,然后油滴破裂释放cDNA,统一进行文库构建,增大了实验通量,但需要专门的实验设备,如BioRad的ddSeq single-cell isolator(用Illumina Nextera kits);Fluidigm C1 platform的低通量的微流控技术,能在捕获细胞时可视化的控制empty wells or doublets。但目前最主流的是10X Genomics。
两种方法的比较如下:


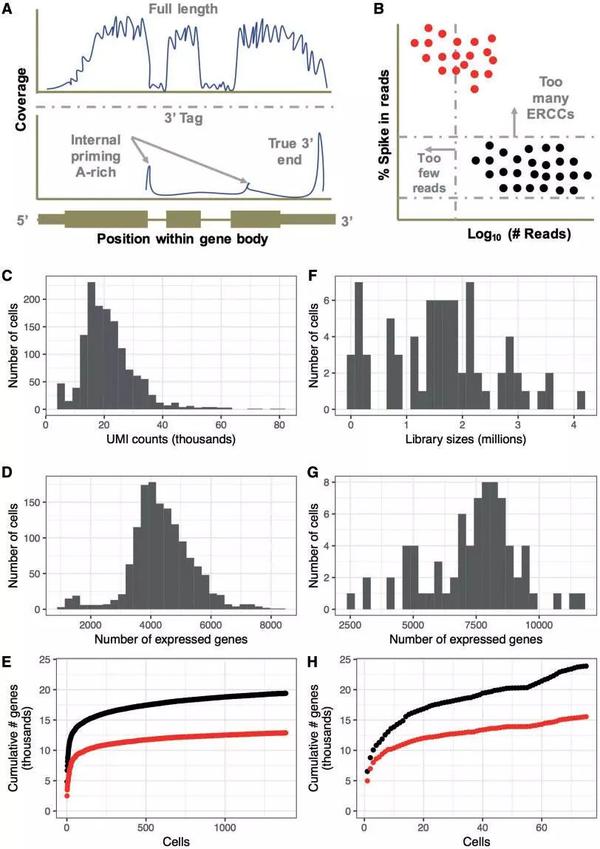


C、D、E:10x data F、G、H:Smart data
(1)从Fig.1A可知,两种方法最大的区别是RNA反转录成cDNA的方法:分别是全长mRNA和3’尾测序,研究表达量较低的RNA可以选择smart的全长测序,而3’端测序可以得到更多的细胞,从而得到一个细胞群体的转录异质性。
(2)spike-in control是常用的评估技术差异的方法, Lun et al.的研究发现spike-in control 在确定测序过程中的empty Wells和的dead cells有重要作用,因为高的ERCC含量与低质量数据相关,并且通常是排除的标准。
Spike-in:A molecule or a set of molecules introduced to the sample in order to calibrate measurements and account for technical variation; commonly used examples include external RNA control consortium (ERCC) controls (Ambion/Thermo Fisher Scientific) and Spike-in RNA variant control mixes。一个分子或一组分子引入到样品中以校准测量并解释技术变化;常用的例子包括外部RNA控制联合体(Ercc)和spike-in RNA变体控制混合物。
(3)10x捕获到的细胞数量更多,实验设计的不同会使文库大小相差几个数量级(Fig.1C,F)
(4)Smart-seq2 可以得到更多的基因信息(Fig.1G,D)
(5)Smart-seq2数据集比10x更复杂(Fig.1E,H)
总之,10x更加方便,简少了人工操作,简化了大量细胞的数据收集。但细胞数的增加导致测序深度也增加,牺牲了文库的复杂度,丢失了固定的工作流程。
03
实验设计
(1)避免技术偏差
成功的测序实验设计 应遵循三个原则:生物学重复、随机分组和平衡区组设计(a balanced block design),见图2.
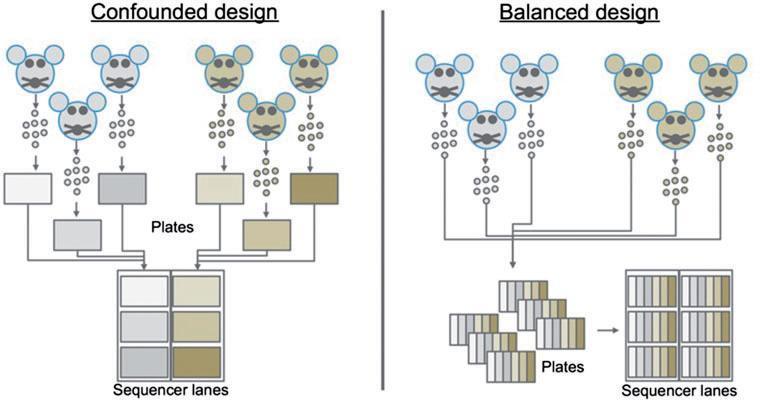



在一个混杂的(confounded design)设计中,来自单个小鼠/条件组的细胞将被分拣到单个板上,并且该板上的细胞文库将在一个批次中制备。最后,在一条lane中测序,而另一个条件下的文库则在另一条lane中测序。在这个例子中,不可能分辨出基因表达的差异是因为实验条件还是混杂的/不均衡的实验设计引入的技术差异。这个实验可以使用平衡组区重新设计,保证每个样本的六个小鼠和测序仪的每一条lane都有样本。
Smart-seq2 允许选择individual wells填充细胞,很容易实现平衡组区的方法。对于10x,需要确保在初始封装过程中处理不同芯片上的样品,并进一步进行下游处理以平衡实验。
(2)决定合适的细胞数目和测序深度
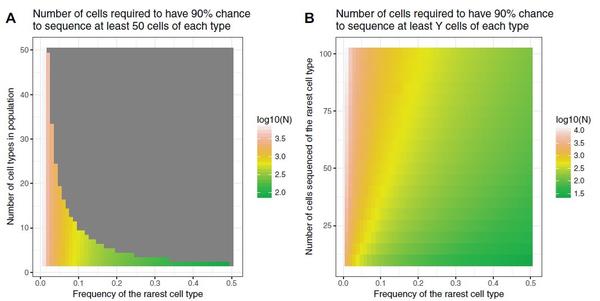



回收细胞数目可以根据样本中所有细胞的预期异质性、样本内特定细胞类型所期望的最小频率和在所得到的数据集中期望的每种类型的细胞的最小数目来估计。可以利用负二项分布评估稀有样本的细胞数目。例如,如果我们要测序好友10个细胞类型的样本,但稀有细胞的频率只有0.03,我们就需要对2200个细胞进行测序,以保证90%的机会捕获到50个稀有细胞。一个实验室:Satija lab,可以提供在线工具(http://www.satijalab.org/howmanycells),基于细胞类型和多样性评估目标细胞数目。
每个实验的测序深度的准确估计需要预知个体细胞中的总mRNA含量和这些细胞中mRNA物种的多样性。但这些参数往往很难预估。在最近发表的研究中提供了一个有用的指导方针,他们用16个不同的protocol和五个物种进行了34个独特实验,对这些单细胞数据进行了全面的分析。他们发现,每个细胞有250 000个reads就足够准确,每个细胞的100万个读取是饱和基因检测的好目标。但具体的测序深度因实验设计差异和物种不同而做适当调整。
END
转载请注明本文转自欧易生物