
Conquer-对单细胞数据差异表达分析的重新审视

随着单细胞测序技术的流行,我们对复杂疾病和性状的理解从patient,tissue的表达谱(bulk RNA-seq)到单个细胞的表达谱(single cell RNA-seq)。究其原因,在于bulk RNA-seq产生的是一个细胞群体的平均读数,而细胞,特别是癌细胞存在极大的异质性,这些平均信号可能不足以反应这堆组织的真实信息。Prof Aviv Regev (MIT教授,HHMI研究院,人类细胞图谱计划项目co-chair)曾经形容这种方法就像水果沙拉,颜色和味道都能提示冰沙的成分,但倘若只有几个是属于蓝莓的味道,那么很容易就被一堆草莓的味道所覆盖(如下图)。因而在细胞尺度上进行大规模的测序分析以对细胞进行重新分型是很有必要的。
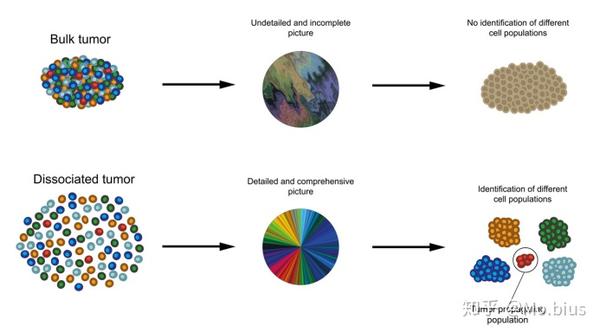

图源:http://fightdipg.org/wp-content/uploads/2015/12/single-cell-sequencing.jpg
然而,类似于传统的在bulk RNA-seq中的分析,我们依然要对两个不同的细胞类群cell type (group)进行差异表达分析(Differential expression analysis),寻找具有显著性表达差异的基因,缩小找marker genes的范围。但是基因在scRNA-seq data中的表达分布并非完全的负二项分布。
一个比较重要的区别是在scRNA-seq Data 中有更多的0值,即“稀疏性(sparse)”。这些0所包含的信息也不一样,受到技术上和细胞本身的两个因素的影响。从技术上讲,大多数mRNA在单个细胞的量是非常少的,而绝大多数基因在一个细胞中只有少量的mRNA的拷贝,由于在mRNA capture protocol的一些问题,可能会导致一些基因的mRNA在逆转录和cDNA扩增阶段存在完全的缺失,进而导致漏测,这种现象被称为dropout event。而也存在一些基因的表达为0的情况是由于其本身在细胞的表达量就非常低,或者由于mRNA bursting的现象(文献可参考:Golding, I; Paulsson, J; Zawilski, SM; Cox, EC (2005). "Real-time kinetics of gene activity in individual bacteria". Cell. 123 (6): 1025–36. doi:10.1016/j.cell.2005.09.031. PMID 16360033)。那么对应的,受这些0的影响,原本的负二项分布(negative binomal distribution)的基因可能更多服从于0膨胀负二项分布(zero-inflated NB)。
基于scRNA-seq数据的特殊性(稀疏,高噪声,高维度),为bulk RNA-seq data设计的差异表达方法是否适合于scRNA-seq data也需要做进一步讨论。但同时即便目前涌现了很多scRNA-seq data,这些data的分析目的不同,使用的pipelines也相差很大,因而重新使用这些已经经过预处理的公开数据,同时进行不同方法的比较也变得很困难。
我们今天主要介绍Charlotte Soneson等于2018年在《Nature Methods》发表的“Bias, robustness and scalability in single-cell differential expression analysis”这篇文章。这项研究利用相同的pipelines处理,整合了多个公开的scRNA-seq数据,同时对所有的注释基因和转录本进行了丰度估计,以及质量评估和研究分析报告提供给使用者以确定他们所需要的合适的数据库。
Conquer为文章作者开发出的一个整合性数据库。目前总共有36个数据集:31个来自于full-length protocols同时5个是3’-end sequencing(UMI) protocol。Conquer对这些数据进行一致的预处理和分析,有利于不同计算方法的比较。同时Conquer还包含了不同的物种和细胞类型可以检验某些生物学假说的泛化性(generality),也就是假说的适用广度和范围。
分析方法/Methods
在这篇文章中,共选择了七个数据集(six full-length and one UMI data)以及两个额外的UMI count数据对如下的差异分析方法进行评估。
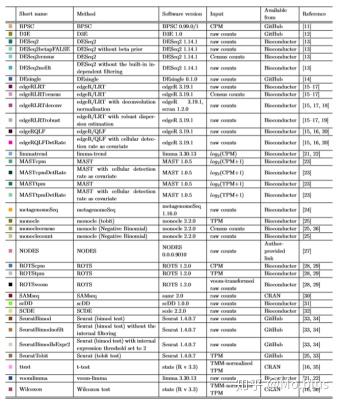
这些差异表达分析方法包括了对bulk RNA-seq data的经典分析方法 (SAMseq, edgeR, limma, DESeq2等), 专门开发出来用于scRNA-seq data的分析方法 (SCDE, DEsingle,Seurat,MAST等)以及一些常见的统计方法 (Wilcoxon, t-test等)
数据准备/data preparation:
Signal data:
我们将之前选择的九个数据库只保留已经注释好的两个cell group,从t-SNE的结果看,在低维流形上绝大多数数据对应的两个cell group在表达上具有明显差异。对于每个数据集,我们生成一个‘maximal’大小的实例 (举例:即每个group的细胞数量等于原始的较小那个cell group的细胞数量),以及从maximal大小的数据集中随机采样(random sampling) 生成若干个小的数据集,这些数据集都作为signal data。
null data:
null data用于后续的type 1 error分析,是在signal data中单个细胞群体中随机采样得到新的数据作为null data。在这些null data中理论上不存在差异表达基因,因为都是来源于一个细胞群体。
Simulation data:
基于powsim R package(链接: https://github.com/bvieth/powsimR)和三个原始数据集生成simulation data进行分析(表示不太懂powsim这个包,不过貌似是生成和原始数据在分布和dispersion等等参数上类似的模拟数据,但选择了特定的10%的基因为差异表达基因),这些simulation data是用于后续的算法性能在TPR和AUROC等等方面进行评估的。
Number of differentially expressed and non-tested genes
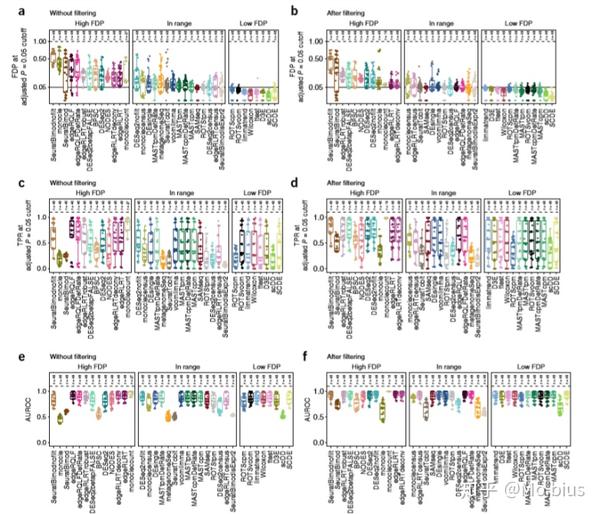
文章对九个scRNA-seq data对应的signal data计算和统计了差异表达基因数量。对于full-length数据集而言,在没有做prefiltering (prefiltering为过度掉低表达基因)的情况下,SeuratBimod检测到了最多存在显著表达差异的基因,而经过prefiltering以后,大多数方法的检测到的差异表达基因数量有所下降,edgeR/QLF检测到的差异表达基因数量下降是最多的。且随着两个group细胞数量的上升,发现的差异表达基因数量越多。
大多数差异表达方法并不是在全部基因上都做了test,除了方法内置有filtering步骤,一些基因也会运行报错,可能是在拟合的步骤无法收敛而去除的一些基因,这在bulk RNA-seq。
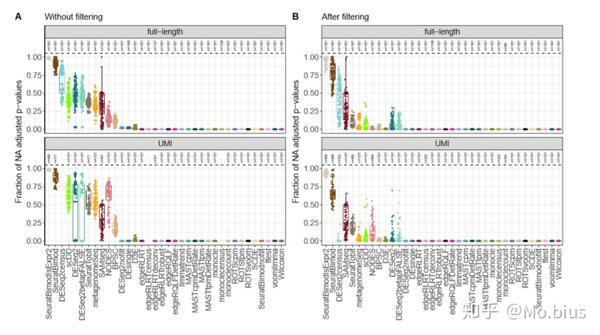

(观察右边的图可以发现相比于左边的没有filtering,返回NA的基因数量明显降低了)
Type 1 error control
Type 1 error也是很重要的,其实这一部分也就是看false positive rate(FPR)在各个数据集各个方法上的情况,对于8个原始的数据中通过对单个细胞组(cell group)进行随机采样得到一个null data,那么在null data中理论上不存在差异表达基因(因为都是取自同一种细胞)。因而我们希望在这些data上运行差异表达分析方法以后返回后的差异表达基因占全部检测的基因数量的占比要小于0.05(对应的就是type 1 error也就是FPR)。
对于unfiltered的数据集而言,ROTS以及SeuratTobit的性能是最好的。但对于edgeR/QLF,SeuratBimod两种方法我们可以看到结果有很多假阳性,经过filtering以后会好很多,但是会少很多检测显著的基因。特别是SeuratBimodIsExpr2返回的DE的基因数量就不是很多
从UMI数据集看,monocle使用TPM数据反而效果不佳,当在full-length数据中,TPM数据的效果比read count要更好,特别是筛选一些低表达基因以后,相比于其他方法, voom-limma,ROTSvoom以及edgeR/QLF这些基于线性模型的bulk 分析方法的性能有了很大提升同时结果更加稳定。
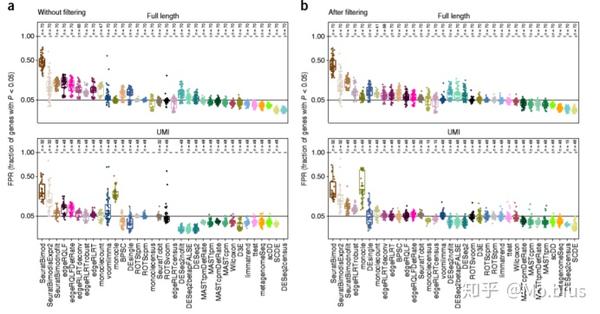

除了FPR,从p value的分布上也有一些很有意思的现象,在没有gene prefiltering的时候,每个方法在p value的分布上并不是均匀的,特别是在SeuratBimod中p value较小的基因占绝大多数,相反的monocle中p value较大的基因占绝大多数。其他基因也存在类似的情况,但经过prefiltering以后都有所改善(趋向于均匀分布),特别的,即便有prefiltering,SeuratBimod依然倾向于给出较小的p value,考虑到之前FPR的结果,SeuratBimod的假阳性就显得有点过高了。
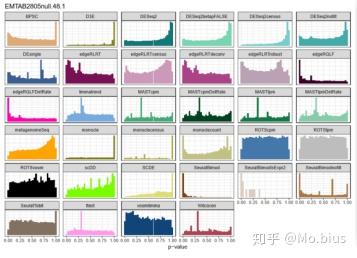
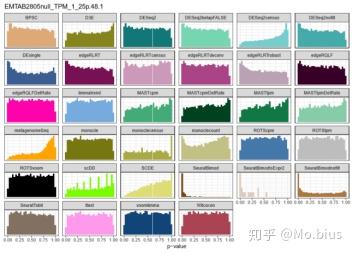
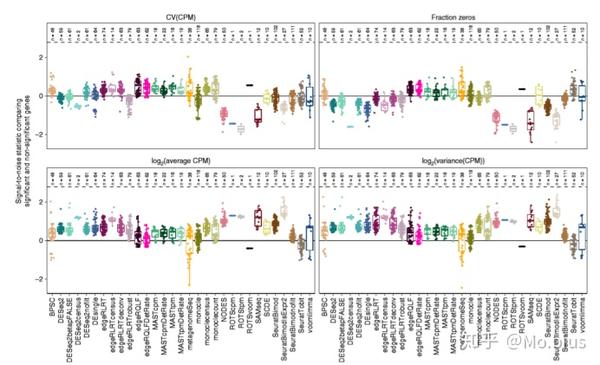
Characteristics of false-positive genes
那么这些false-positive genes和其他True-negative genes在表达的特征上有什么区别?这个研究主要估计了各个基因的四个表征(characteristic):平均表达量(average of CPM),方差(variance of CPM),变异系数(variance and coefficient of variantion of CPM)以及表达量为0的细胞在所有细胞中的占比(Fraction Zeros)。同时计算了信噪比统计量进行比较(signal-to-noise,其实可以理解为scaling,方便不同方法之间进行比较)。结果发现对于NODEs,ROTS,SAMseq,SeuratBimod这些方法而言,假阳性的基因表达量为0的情况比较少,同时表达量较高,变异系数较小。相反的,对于edgeR/QLF,SeuratTobit,MAST以及metagenomeSeq有较多的表达量为0的值,平均表达量较小,变异系数较大。

Between-method similarity
对于一个差异表达分析方法而言,可重复性是很重要的,因而每次分析出的差异基因的一致性(concordance)不应受到样本量的影响。或者说,如果对于两个population中有真实的潜在的差异信号,不管取这两个population中多大的样本量我们都可以发现这个signal。因而假如我们随机抽取样本做差异表达分析,每次做出的结果应该有很高的一致性。相反的,对于两个population中不存在差异表达信号的(null data),我们每次做出的结果理应是随机的,不一致的,倘若存在一致性。那么这个数据集或者方法或者说两者的组合就会存在一定的bias。
因而基于此,为了测量(measure)这种一致性,我们可以依据显著性(pvalue)进行rank,我们选择top100个具有显著性差异的基因,在每一对具有相同样本量的数据实例(data instance)的比较中,我们看top1-100有多少个基因是一致的,比如Top20个基因中有五个基因是一样的,那么就记为5。从横坐标1-100对应的作出纵坐标相应的一致基因的数量,就可以得到一条一致性曲线(concordance curve),类似于AUC之于ROC,我们计算AUCC,也就是一致曲线的下面积。同时除以100^2/2进行标准化(100^2/2相当于从top1-100都是一致的)。
从结果上看,我们可以看到显著的signal data中存在较高的similarity(concordance),相反的null data的一致性就不是很高了。一个良好的数据集在signal data与null data中一致性差异应该是比较大的,而EMTAB2805这个数据集signal data与null data差异却不是很高,而10XMonoCytoT的差异就很明显了。有意思的是,一些无参数方法(Wilcoxon test,SAMseq)以及一些log-like transformation处理的方法(limma-trend, ROTSvoom, voom-limma)相比于基于count数据分析的方法在signal data和null data之间的差异更大,说明在鲁棒性上,对于scRNA-seq数据的一些小的变化,基于count数据可能更加敏感。
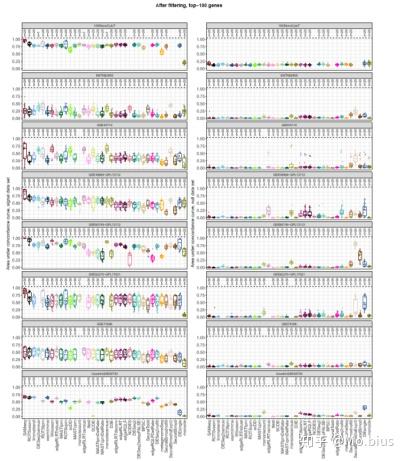
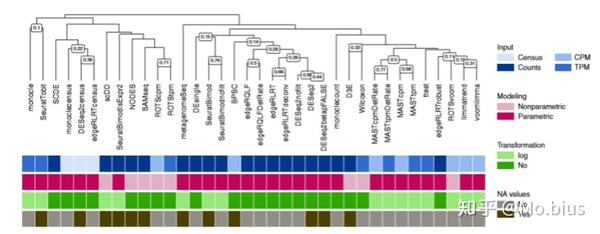

利用AUCC,我们也可以比较不同方法之间结果的相似性,从聚类的结果可以看出来,依据数据的输入格式,transformation的方式以及modeling的方式,都会对方法的一致性产生影响。但同时需要指出的是,如果在进行一致性比较时两个实例中的细胞数量与AUCC值存在一定的相关性。
FDR control and power
最后,这项研究也基于simulated data比较了FDR control,TPR(true positive rate)以及AUC的情况。一些方法如基于Census data的voom/limma,ROTSpm,MAST,以及有prefiltering的SeuratTobit,SeuratBimod和SAMseq都能控制FDR在0.05这个水平左右,而SCDE,scDD和t test,limma-trend以及wilcoxon test等等方法可以控制FDR低于0.05这个水平。edgeR/QLF的FDR水平较高,经过filtering以后,edgeR/QLF的FDR显著下降。同时大多数方法提高样本量都能降低FDR的水平。
从真阳性率TPR上去看,经过filtering后,如edgeR/QLF,SAMseq,DEsingle还有voom-limma均有良好的表现。DESeq2在不管有无intern filtering后均有良好的表现,同时从AUROC上去看的话,最好的是edgeR,MAST,limma以及SCDE等等,经过prefiltering后,大多数方法的AUROC值都有了相对的提高。文章最后还比较了各个算法的运行速度和这些方法的开源性以及documentation,不作详细的介绍。

结论
从这篇研究我们可以看到,过去一些研究认为的在bulk RNA-seq中分析的方法不太适用于scRNA-seq数据,究其原因在于scRNA-seq由于Drop-out event以及单个细胞的transcript太少,估计出的单个基因在多个细胞中的表达水平会存在大量的0值,因而这时候的基因应该服从0膨胀的负二项分布分布(zero-inflated negative binomial distribution,可以理解为在0这一点的狄拉克分布和负二项分布的混合分布),不同于在bulk RNA-seq中的负二项分布。
而这一研究通过对不同方法在不同数据以及不同输入格式(CPM, TPM, Census Count等)进行比较,发现prefiltering确实会显著影响到DE算法的性能,同时传统的一些limma,edgeR在scRNA-seq data中一样能有很好的表现。但值得注意的是,如edgeR和limma对一些0值较多的表达量比较低的基因检验显著,因而在prefiltering这一步相比于bulk RNA-seq要更加严格。
其实懂得No Free Lunch theory(1、一种算法(算法A)在特定数据集上的表现优于另一种算法(算法B)的同时,一定伴随着算法A在另外某一个特定的数据集上有着不如算法B的表现;2、具体问题具体分析。)便会知道,即便我们看到了edgeR和limma这些在bulk RNA-seq中较好的软件可能在scRNA-seq一样也适用,但是依然得从数据的分布出发,假如我们手头的scRNA-seq数据绝大多数基因表达都比较稀疏,其实使用有prefiltering的一些方法可能会错过很多signal。因而我们可以画出 scRNA-seq data整体表达情况的分布图,同时从分布上去看,是否服从0膨胀分布的基因的占比也会影响到算法的robust也是值得讨论的。
除了在方法的选择上看,对于今后凡是做算法性能比较的real data(在做出新的算法时为了和过去算法进行比较,需要真实的数据进行性能比较),这项研究也给出了很有意义的参考。我们可以看到一些scRNA-seq data分析出的差异表达基因的可重复性容易受到比较的时候细胞数量的影响,而选择更稳定的,受随机因素影响更小的scRNA-seq data也许在与其他算法进行比较时会更好,比如10XMonoCytoT。为了给用户提供适合他们使用目的的scRNA-seq数据,conquer还提供了探索性分析报告。该报告计算并可视化细胞的各种质量测量,并提供细胞的低维表示,由不同的表型注释着色等等信息。
(http://imlspenticton.uzh.ch:3838/conquer/)
同时,在单细胞测序数据分析中,不仅仅在差异表达分析上存在一些需要讨论的问题,在做依据表达谱进行细胞分型也是需要做进一步标准化数据库,方法等等,放在同一个框架下讨论各个方法的适用范围。之前听的一个seminar上就说目前几十种聚类算法,能找到几十种不同的分型结果,富集到各种不同的pathways,究竟哪一种更make sense;以及,在bulk RNA seq data中用于聚类的经典算法(PCA, K-means等)能否一样适用于scRNA-seq data,可能也要像这项研究一样进行更加细致的讨论。
参考文献:
[1] :Soneson C, Robinson M D. Bias, robustness and scalability in single-cell differential expression analysis[J]. Nature Methods, 2018.
[2] Miao Z, Deng K, Wang X, et al. DEsingle for detecting three types of differential expression in single-cell RNA-seq data.[J]. Bioinformatics, 2018.
