生信log19|原核基因组分析part2-本地个性化分析(序列建库部分真核也适用)

这篇写得太久了,还有很多不满意的地方,决定先放出来再慢慢完善。 刚来实验室的那会儿,经常听到实验室的师兄说建库,pfam,antismash诸如此类的名词,我听得一头雾水,处于想学但是学不明白,只能暂时把这些东西记在脑海中。更别说实践起来的时候,各种坑都踩了个遍,诸如此类分析的流程和标准,以及为什么要这样做,当时也没能解决。写这篇的目的在于记录找新基因或者是个性化基因的本地自建库流程以及当时迷惑的点,也供其他小白同志们参考一下,这种方法也适用于真核生物的建库。
1、为什么要自建数据库?
下面列出现场数据库的几宗“罪” * a. 检索的困难:公共数据库存放了全世界各地的数据,信息量非常大,没有大海捞针般困难,批量检索数据的时候,要花费的时间和精力也是不少的,时长从几分钟到一个星期不等,十分考验计算机的算力,使用网页版的吧,批量处理不友好,而且大部分网页工具现在询问的条数,上传文件数量的限制(以KEGG为例大概是5000条蛋白序列,每次提交的文件,限每个邮箱一个);
- b. 针对性问题:数据库收录序列广了未必就是好事,实际操作中不但速度慢,而且目标太多会干扰比对结果,非常有可能比对不上自己想要的同源序列而是比对到了其他非同源序列。
- c. 收录的问题:第一点,一些数据库的历史比较悠久,像收录16S rRNA基因的Greengene数据库,RDP数据库还有直系同源蛋白质分类COG数据库已经不再更新,相信还有更多的数据库已经停止更新。第二,一些数据库到目前为止还有维护和更新,但由于信息需要验证,速度跟不上文献发表的速度,因此最新的序列需要通过搜查文献才能得知。
总结一下,个性化建立数据库的是为了更加高效,批量化地与特定范围的同源序列进行比对。
常用蛋白质数据库的完整度和可信度对比
完整度:NCBI > Uniprot > Swissprot
准确度:Swissprot > Uniprot > NCBI
PS:这里并没有讨论 Pfam的数据库是因为pfam建库的依据是蛋白质的结构域序列建库的。另外NCBI的数据库的更新速度应该是最快的,因为发文章之前统一都需要把数据上传到Genbank或者EMBL数据获得Accession number才能
2、数据库到底是长成什么样的
组成数据库的元素:核苷酸序列,氨基酸序列等等,只要是同类的材料并且数量多都可以组成数据库。这里只讨论核苷酸序列和氨基酸序列的数据库,并侧重于氨基酸序列数据库。下面展示三种不同算法的建库后的文件


3、三种常用建立数据库的方法及结果解析
下面几种建库方法是建立在蛋白质序列或者核苷酸序列基础上的,建库的方法其实也就是背后的模型不一样,一个是动态规划算法,一个是diamond算法,一个是隐马尔可夫模型。这些预测模型被打包成软件,所以我们在使用的时候并不复杂。


个性化搜索的步骤
0、多序列比对(此步为Hmmer算法必须的,Blast和diamond不需要) * 多序列比对可用clustalw或者是mafft
# mafft 比对
mafft --auto in_protein.fa > out.fa
# clustalw 用这条命令时,把Protein换成自己的蛋白的名称
echo "1\nprotein.fa\n2\n1\nProtein.aln\nprotein/dnd\nX\n\n\nX\n |clustalw"1、建库 2、根据自己建的数据库搜索未知序列 3、结果提取和数据验证 4、可视化
3.1 Blastp算法建库数据库
# 建库
makeblastdb -in 用来建库的核苷酸/氨基酸.fasta \
-dbtype nucl/prot \ #选择数据的类型(蛋白质还是核苷酸,上面一样)
-parse_seqids \ #这个是要保留原来序列 ">"后面跟着的字符串
-out yourdb_name
# 搜索
blastp -db database_name \
-query your_file.faa -outfmt 6 \
-out output_filename \
-evalue 1e-5补充说明一下上面的参数-parse_seqids是用来保留原来序列的identifier的信息的就是“>”后跟着的序列名称。另外输出的结果均选择格式6
3.2 Diamond多序列比对方法建库
#建库
diamond makedb --in 同源序列文件 --db nr
# 搜索 blastx是给DNA序列用的,blastp是给蛋白质序列用的
diamond blastx --db nr -q reads.fna -o dna_matches_fmt6.txt
diamond blastp --db nr -q reads.faa -o protein_matches_fmt6.txt3.3 HMM算法建库
#构建模型,使用多序列比对的结果文件
hmmbuild filename.hmm 多序列比对的输出文件.fasta
#搜索
hmmsearch --domtblout out_protein.txt --cut_tc protein.hmm query_file.faaPS:数据库的命名建议用跟同源序列相同的名字,以便后面辨认
三种建库方法的评价及阈值设定问题
blastp的算法是最经典的至今大量文献仍然使用这个算法尤其是分子生物学,一般在blastp结果中,相似性为30%的序列都能被人认为具有一定的生物学功能相似性,相似性在70%或以上则被认为是具有相同功能的蛋白质或者是完全一样的蛋白类型,并且实验室验证也很容易获得相同的活性结果。Blastp做序列比对中目前还是必要的选项,但是在大批量作业下速度太慢,对发现潜在性新蛋白不敏感。diamond:跟楼上的blastp算法结果比较相近,但胜在速度快,据其文章所言,序列越多比对速度越快,单序列与多序列想比对,速度反而慢。Hmmer:隐马尔可夫模型是根据氨基酸的频率来预测新的序列,依据是蛋白质结构域,速度非常快,利于找到新的序列信息,美中不足的是找的并非是功能蛋白的序列全长,因此分子实验可能会不太能验证。
3、数据解析和信息提取(fasta序列提取)
- 用linux三件套(grep,sed,awk)提取相应的信息 附正则表达式命令行提取文本中的
KO号(提取方法不唯一,我的也不一定具有应用普遍性),注意到KO号实际上的组成是字母K后面再跟一串数字,但数字有多长我们并不清楚
#用grep提取从KEGG上注释到的KO号
cat 45_upload.txt|grep -h 'K[0-9].*[0-9]' > final_result.txt

blastp结果的提取
- blast的结果选择的是格式6,默认的行数有12列,每一列的内容如表格所示(可以有目的地筛选所需要的信息);
| qseqid | sseqid | pident | length | mismatch | gapopen | qstart | qend | sstart | send | evalue | bitscore | | ------ | ------ | ------ | ------ | -------- | ------- | ------ | ---- | ------ | ---- | ------ | -------- |
1. qseqid :查询的id(我们的序列号);2. sseqid :数据库中的参考序列的序列号;3. pident :匹配到的概率;4. length :序列比对上的长度;5. mismatch number of mismatches:错误匹配的概率;6. gapopen number of gap openings:间隔的数目;7. qstart start of alignment in query:查询序列的起始位点;8. qend end of alignment in query:查询序列的终止位点;9. sstart start of alignment in subject:参考序列的起始位点;10. send end of alignment in subject:参考序列的比对结束位点;11. evalue expect value:匹配结果出错的概率;12. bitscore bit score:序列得分
- 命令中的
$3表示的是列,如此类推,$4是第四列,$11是第十一列,选取e-value小于0.1的五次方的结果,相似度(概率大于30%)的,以及序列长度选择大于100个氨基酸长度的结果。
cat blastp_output.out | \
awk '{if ($3>35 && $11<0.00001 && $4>100) print $1','$2','$3','$11','$12}' \
> final_result.txtHmmer结果
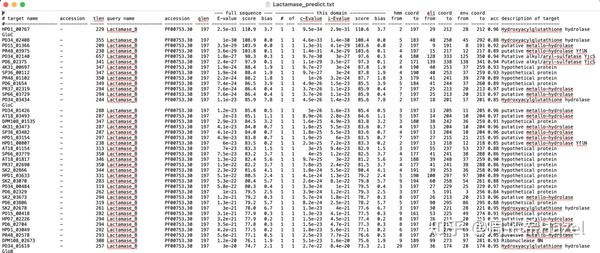

* 建议可以使用excel表中的分列功能,然后把序列的id提取出来,再把原来的查询的序列文件中的对应序列抽取出来。
4、预测数据的验证
在此解释一下为何要再做生物信息学的验证,而不是直接上手分子生物学的验证。我们下载到的序列不一定都是全是“对的”,这除了数据库信息中数据命名混乱以外,我们非常有可能下载到的序列并不是真正想要,可能只是近似的序列,得到一个接近的假阳性结果,因此我们需要在得到一个简化结果之后再与大的数据库进行比对来排除下载对应序列的错误。
以下几个验证的数据库都会互相联动,比如说pfam的数据库可以同时☑️勾选SMART,CDD这些数据库 hmmer search-pfam SMART结构域数据库:做分子的博士后说这个数据库比较靠谱







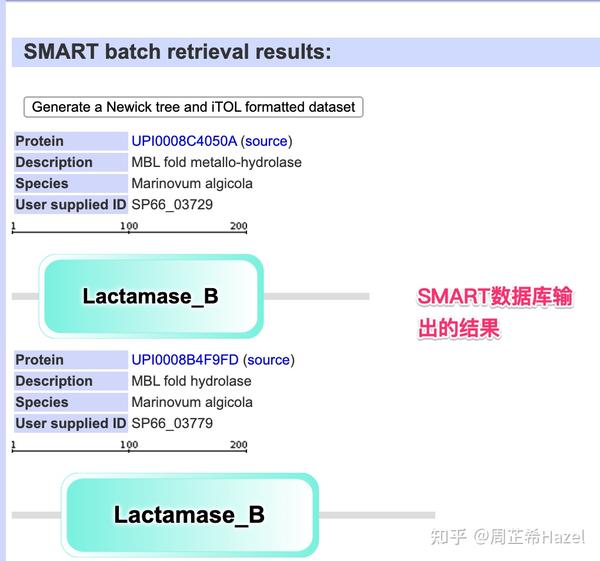

- smart 会可视化出来结构
NCBI的保守结构域数据库验证
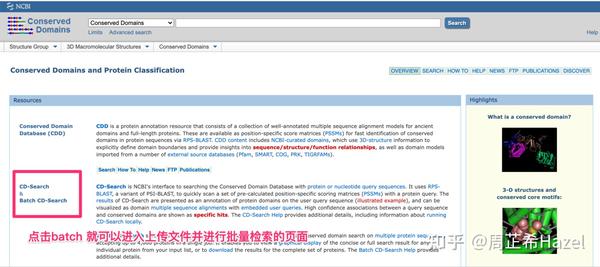



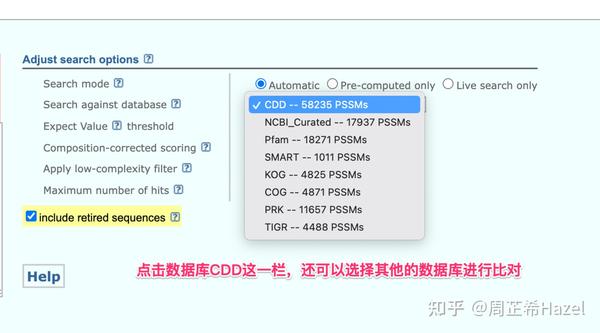

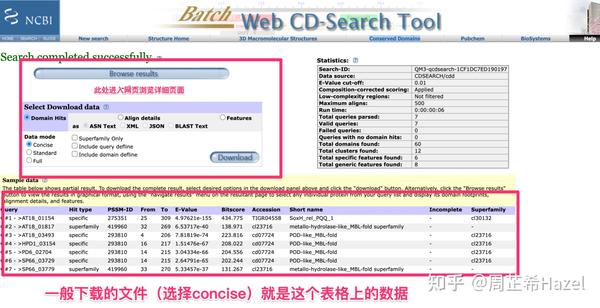





Pfam数据库验证








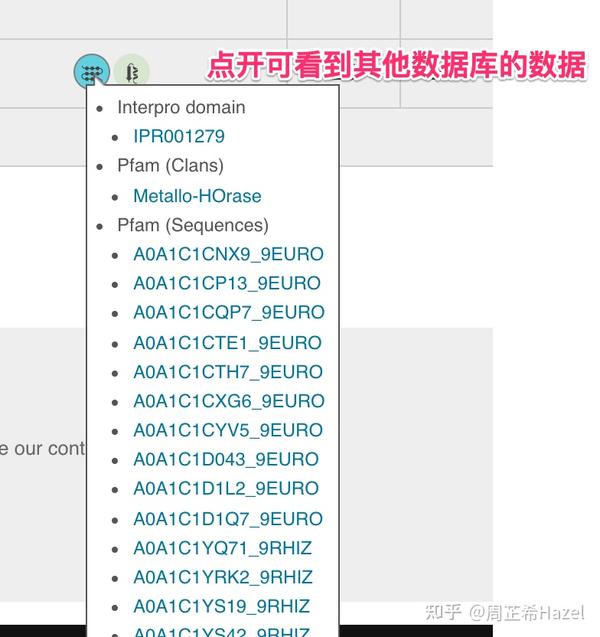

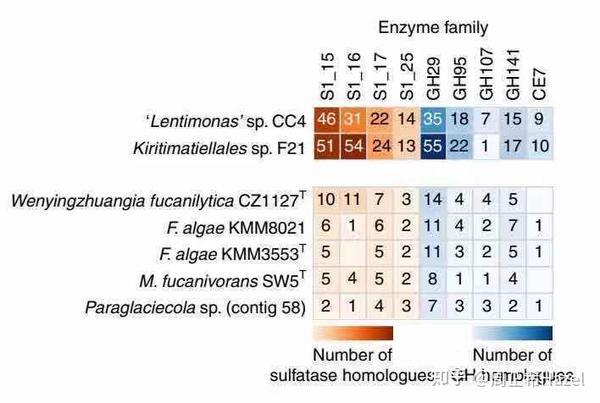
上面的表格数据数据可视化-展示结果事例参考
- 通路重构 KEGG通路重新构建


- 热图

- 序列比对图


- 基因组结构展示圈图 基因结构的展示


- 基因簇结构可视化(gggene)


参考和延伸阅读
- PS:一篇文章实在呈不下太多,此篇仅展示主体部分,里面没有深入的点和可视化的部分以后都会一一在其他log中记录,各位看官有兴趣走过路过点个关注支持留意一下! 有疑惑和指教请在下方留言区留言一起交流学习哦。此篇到这里已经全部更新完毕。
