
基因集富集分析(Gene Set Enrichment Analysis, GSEA)


下面这篇文章中简单介绍了基因矩阵转置文件(gmt)
而下面这篇文章展示了如何使用R来读取gmt格式的文件
今天我们来看看如何做GSEA(Gene Set Enrichment Analysis,基因集富集分析)以及GSEA的结果如何解读。
首先我们需要了解一下GSEA跟传统的基因富集分析有什么区别,有什么优势。我相信大家在做传统的基因功能富集分析的时候肯定遇到这样的情况,一条富集到的通路中,既有上调的差异表达基因,也有下调的差异表达基因,那么这条通路总体是被抑制还是被激活呢?那么这条通路中的基因表达水平在实验组相比于对照组究竟是上升了呢,还是下降了呢?
在传统的富集分析时,我们其实根本不关心这些差异表达的基因究竟是上调还是下调。这是因为传统的富集分析根本不考虑基因表达量的变化趋势,其算法的核心只关注这些差异表达基因的分布是否跟随机抽样得到的分布一致,即使在后续可视化时,我们在通路图上用不同颜色标记了上调和下调的基因,但是由于没有采用有效的统计学方法去分析这条通路中所有差异基因的总体变化趋势,这使得传统的富集分析结果无法回答上述的问题。
即使有些文章里面根据差异表达基因的上下调将差异表达基因分成两组分别进行基因富集分析,这样得到的结果也会有失偏颇,并不能反应差异表达基因的整体情况。有时甚至会出现自相矛盾的情况,上调的基因和下调的基因富集到相同的一条通路中,这时就很难解释结果了。
GSEA(Gene Set Enrichment Analysis),该方法发表于2005年的Gene set enrichment analysis: a knowledge-based approach forinterpreting genome-wide expression profiles,是一种基于基因集的富集分析方法,在对基因表达数据分析时,首先确定分析的目的,即选择MSigDB中的一个或多个功能基因集进行分析(基因矩阵转置文件格式(* .gmt)中已经介绍过),然后基于基因表达数据与表型的关联度(也可以理解为表达量的变化)的大小进行排序。然后判断每个基因集内的基因是否富集于表型相关度排序后基因列表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。以上其实就是GSEA的分析原理。下面我们就借助一张图来帮助大家更好的理解GSEA的分析原理。


GSEA的输入是一个基因表达量矩阵,其中的样本分成了A和B两组,找到两组之间差异表达的基因,然后根据foldchange进行排序,用来表示基因在两组间表达量的变化趋势。排序之后的基因列表其顶部可以看做是上调的差异基因,其底部是下调的差异基因。GSEA分析的是一个基因集下的所有基因是否在这个排序列表的顶部或者底部富集,如果在顶部富集,我们可以说,从总体上看,该基因集是上调趋势,反之,如果在底部富集,则是下调趋势。
以上就是GSEA的分析原理,那么进行GSEA的结果怎样解读呢?
GSEA分析结果最常见的是下图:
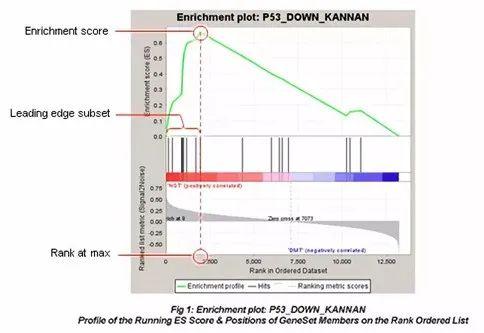

1、图最上面部分展示的是富集分数(ES,enrichment score)值计算过程,从左至右每到一个基因,计算出一个ES值,连成线。在最左侧或最右侧有一个特别明显的峰值就是基因集表型上的ES值。图中间部分每一条线代表基因集中的一个基因,及其在基因列表中的排序位置。
2、最下面部分展示的是基因与表型关联的矩阵,红色为与第一个表型(class A)正相关,在class A中表达高,蓝色与第二个表型(class B)正相关,在class B中表达高。
3、Leading-edge subset 对富集得分贡献最大的基因成员。若富集得分为正值,则是峰左侧的基因;若富集得分为负值,则是峰右侧的基因。
4、FDR GSEA默认提供所有的分析结果,并且设定FDR<0.25为可信的富集,最可能获得有功能研究价值的结果。但如果样品数目少,而且选择了gene_set作为Permumation type则需要使用更为严格的标准,比如FDR<0.05。
下面我们来看看如何使用R语言来进行GSEA分析,这里跟大家分享两种方法,一个是fgsea包,另一个是clusterProfiler包。
一、fgsea包,fgsea是Fast Gene Set Enrichment Analysis的缩写
1. 首先需要准备好rank文件,就是排好序的基因列表文件。一般做完差异表达分析都能得到这样一个文件。第一列是entrez gene id号,第二列可以是t值,也可以是foldchange。
> ranks
ID t
1 170942 -63.337034
2 109711 -49.747787
3 18124 -43.638784
4 12775 -41.518887
5 72148 -33.260388
6 16010 -32.776263
7 11931 -29.423282
8 13849 -28.834755
9 241230 -28.651111
10 665113 -27.815833
11 18619 -27.495304
12 21414 -26.938387
13 16194 -26.812283
14 12549 -25.884426
15 78286 -25.103614
16 623121 -25.007892
17 56489 -24.697156
18 217410 -23.698774
19 17992 -23.309611
20 67298 -23.153141
21 16763 -23.008225
22 18197 -22.970447
23 71994 -22.524729
24 235633 -21.937764
25 76566 -21.906042
26 74646 -21.884332
27 330814 -21.605632
28 12517 -21.542185
29 13542 -21.380340
30 70292 -21.0010662. 同时还需要准备gmt文件,gmt文件的下载我们在基因矩阵转置文件格式(* .gmt)一文中已经讲解过,读入到R之后如下
> head(lapply(pathways, head))
$`1221633_Meiotic_Synapsis`
[1] "12189" "13006" "15077" "15078" "15270" "15512"
$`1368092_Rora_activates_gene_expression`
[1] "11865" "12753" "12894" "18143" "19017" "19883"
$`1368110_Bmal1:Clock,Npas2_activates_circadian_gene_expression`
[1] "11865" "11998" "12753" "12952" "12953" "13170"
$`1445146_Translocation_of_Glut4_to_the_Plasma_Membrane`
[1] "11461" "11465" "11651" "11652" "12313" "12314"
$`186574_Endocrine-committed_Ngn3+_progenitor_cells`
[1] "18012" "18088" "18506" "53626"
$`186589_Late_stage_branching_morphogenesis_pancreatic_bud_precursor_cells`
[1] "11925" "15205" "21410" "246086"
3. 看每一套基因集中的基因是否显著富集在差异表达基因列表的上部或者下部
#进行富集分析
fgseaRes <- fgsea(pathways, ranks, minSize=15, maxSize=500, nperm=1000)4. 显示显著富集在上部和下部的各10条通路的NES,pval和padj
由于ES是根据分析的数据集中的gene是否在一个功能gene set中出现来计算的,但各个功能gene set中包含的gene数目不同,且不同功能gene set与data之间的相关性也不同,因此,比较data set在不同功能gene set中的富集程度要对ES进行标准化处理,也就是NES
NES=某一功能gene set的ES/数据集所有随机组合得到的ES平均值


5. 柱形图展示显著富集在上部和下部的各20条通路的富集分值


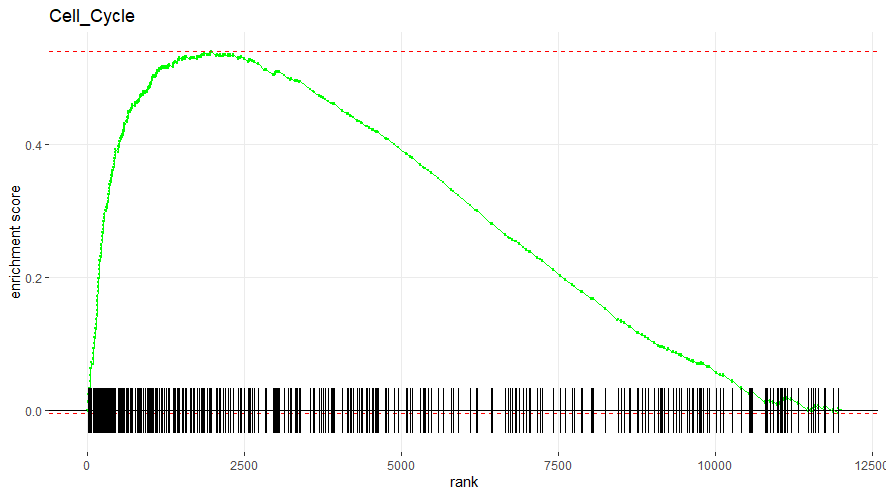
6. 显示特定通路上的富集结果
显著富集在cell cycle通路上部

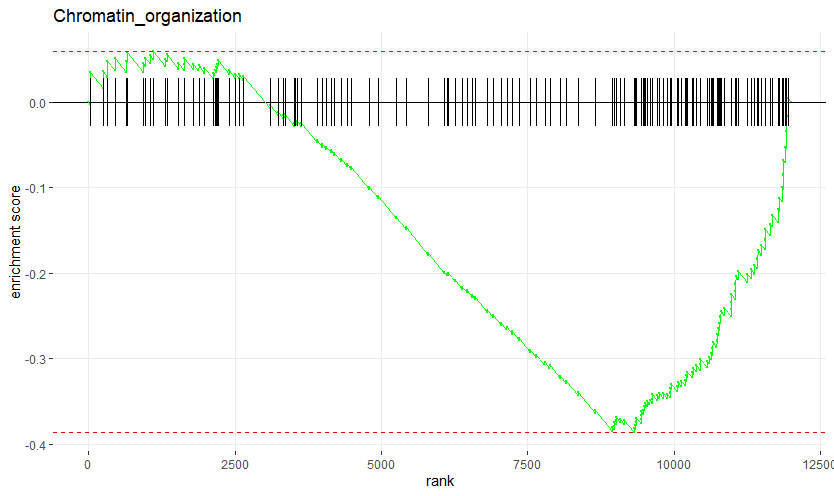
显著富集在chromation organization通路下部

二、clusterProfiler包
步骤跟fgsea包差不多
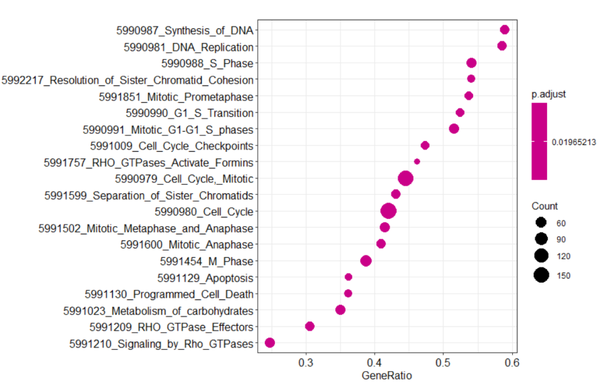
气泡图展示显著富集的前20条通路
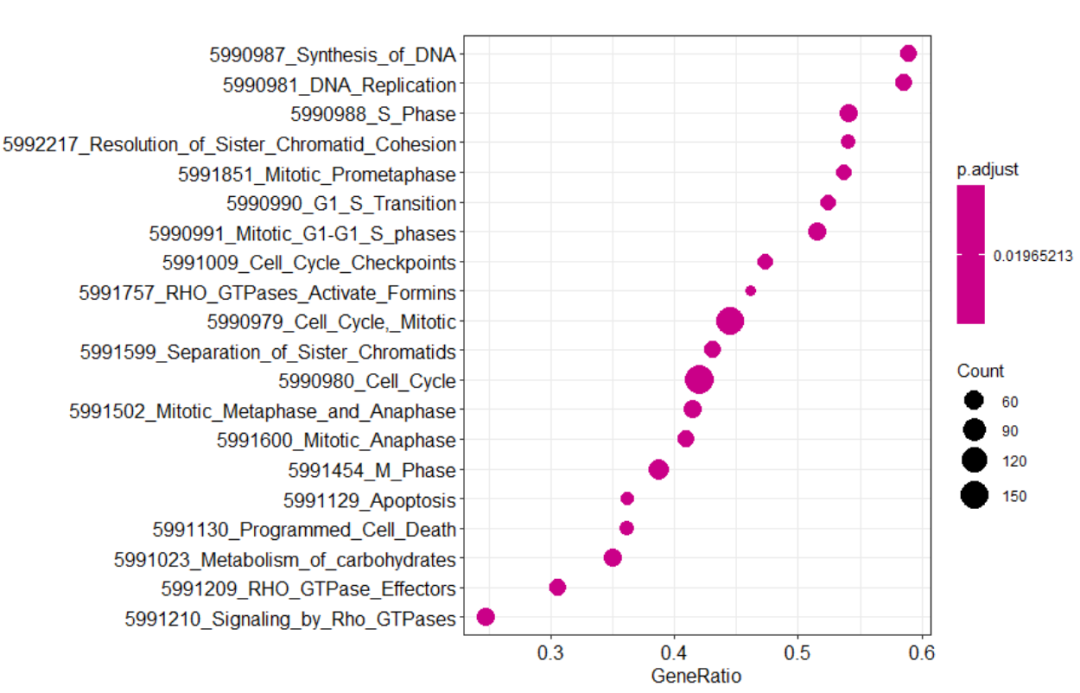
绘制cell cycle通路,显著富集在上部


具体的R脚本参考下面这篇文章

