
HUMAnN 3.0 (alpha)安装及使用

知乎对markdown兼容不佳,可以到csdn查看内容https://blog.csdn.net/weixin_42072765/article/details/108357644
[toc]
简介
(由于humann3和humann2原理是一致的,所以了解huamnn2的原理就可以了。简介内容来自刘大神的一篇博文,是humann2的文章介绍) HUMAnN2是一款快速获得宏基因组、宏转录组物种和功能组成的软件。 与传统的翻译比对方法相比,采用分层式(tiered)检索策略,可以在环境和宿主相关群体中快速、准确获得种水平的功能组成。 HUMAnN2采用比对泛基因组的方法鉴定群体的已知物种,并进一步翻译检索末分类的序列,最终定量基因家族和通路。 结果同时获得功能通路中具体物种组成,建立起了物种与功能的联系,可进一步研究功能组成的贡献者。 应用HUMAnN2研究海洋代谢和生态贡献模式的阶梯变异(clinal variation),实现了人类微生物组通路、物种基因组变异与转录贡献和株水平组成的分析。 作者引入了贡献多样性的概念(contributional diversity),以解释不同微生物群体类型生态学组装的模式。 * 软件特点:
1. 可对已知和末知生物分析群体功能谱
-- MetaPhlAn2和ChocoPhlAn泛基因组数据库, 可以更快速准确获得功能谱 -- 物种包括古菌、细菌、真核生物和病毒 2. 可获得基因组、基因和通路层面的结果 -- UniRef数据库提供基因家族的定义 -- MetaCyc通路基因通路的定义 -- MinPath提供定义的最小通路集 3. 简单的使用界面(单行命令工作流) -- 用户只需提供质控的宏基因组或宏转录组数据 4. 加速序列比对 -- 采用Bowtie2加速核酸水平搜索 -- 采用Diamond加速翻译蛋白水平搜索
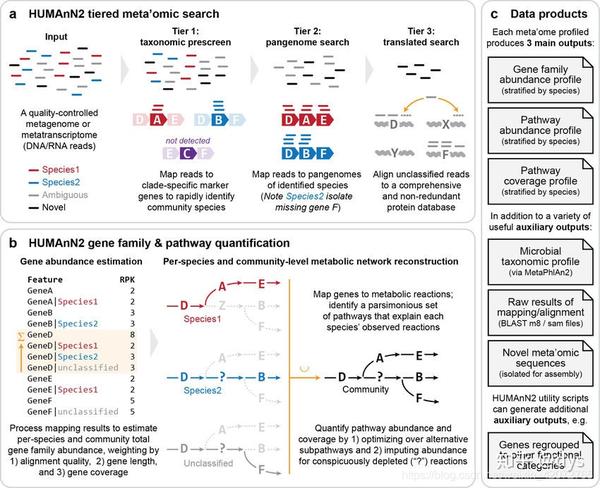
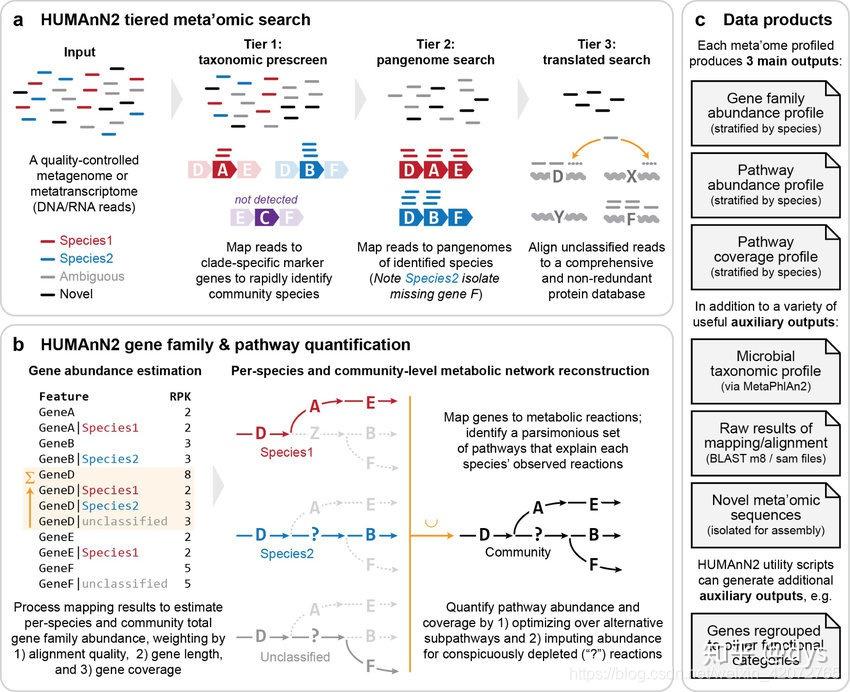
Humann2工作流程
安装
安装方式有多种,个人还是比较推荐使用conda虚拟环境安装。假设之前有安装了metaplan3,可以在metaphlan3的虚拟环境中继续安装,直接conda安装即可。如果没有安装metaphlan3的,可以新建一个虚拟环境,在虚拟环境中直接安装humann3,会自动把metaphlan3也安装上。因为metaphlan3是humann3中的一个依赖软件。可以参考上一篇关于metaphlan3的文章。MetaPhlAn3安装及使用。metaphlan2和humann2都是使用python2的,而metaphlan3和humann3都是使用python3,所以要匹配一起安装,不然依赖关系容易出错。
## 构建虚拟环境,并安装python3.7
conda create --name mpa -c bioconda python=3.7
## conda安装humann
source activate mpa
conda install humann
## 安装完成后,可以运行下面命令测试是否安装成功。
humann_test数据库下载
安装完成后,需要下载humann数据库。最主要的三个数据库,chocophlan 泛基因数据库,uniref 基因家族数据库,utility_mapping 注释数据库。
humann_databases --download chocophlan full /path/to/databases --update-config yes
humann_databases --download uniref uniref90_diamond /path/to/databases --update-config yes
humann_databases --download utility_mapping full /path/to/databases --update-config yes使用humann_databases 命令可以查看数据库下载地址。下载太慢,建议直接网页下载后再上传到/path/to/databases(个人具体路径)
chocophlan : full = http://huttenhower.sph.harvard.edu/humann2_data/chocophlan/full_chocophlan.v296_201901.tar.gz
chocophlan : DEMO = http://huttenhower.sph.harvard.edu/humann2_data/chocophlan/DEMO_chocophlan.v296_201901.tar.gz
uniref : uniref50_diamond = http://huttenhower.sph.harvard.edu/humann2_data/uniprot/uniref_annotated/uniref50_annotated_v201901.tar.gz
uniref : uniref90_diamond = http://huttenhower.sph.harvard.edu/humann2_data/uniprot/uniref_annotated/uniref90_annotated_v201901.tar.gz
uniref : uniref50_ec_filtered_diamond = http://huttenhower.sph.harvard.edu/humann2_data/uniprot/uniref_ec_filtered/uniref50_ec_filtered_201901.tar.gz
uniref : uniref90_ec_filtered_diamond = http://huttenhower.sph.harvard.edu/humann2_data/uniprot/uniref_ec_filtered/uniref90_ec_filtered_201901.tar.gz
uniref : DEMO_diamond = http://huttenhower.sph.harvard.edu/humann2_data/uniprot/uniref_annotated/uniref90_DEMO_diamond_v201901.tar.gz
utility_mapping : full = http://huttenhower.sph.harvard.edu/humann2_data/full_mapping_v201901.tar.gz上传完成后,解压数据库压缩文件
mkdir chocophlan_v296_201901
mkdir uniref90_v201901
mkdir mapping_v201901
tar -zxvf full_chocophlan.v296_201901.tar.gz -C ./chocophlan_v296_201901/
tar -zxvf uniref90_annotated_v201901.tar.gz -C uniref90_v201901
tar -zxvf full_mapping_v201901.tar.gz -C ./mapping_v201901/更新config文件,主要是更新数据库路径,其他的设置更新也是可以通过这种方式更新
## 更新格式:humann_config --update <section> <name> <value>
humann_config --update database_folders nucleotide /path/to/databases/chocophlan_v296_201901
humann_config --update database_folders protein /path/to/databases/uniref90_v201901/
humann_config --update database_folders utility_mapping /path/to/databases/mapping_v201901/
## 更新后查看设置
humann_config运行
humann3和humann2的运行命令基本没有差别,就是把huamnn2换成huamnn,说人话就是把脚本名中的版本号去除。其他运行脚本名也是同样处理。
## 运行必须参数,输入文件及输出路径。
## 输入文件可以为多种数据形式,包括测序文件及压缩格(fasta, fastq, fasta.gz, fastq,gz) , 比对文件(sam, bam等),还有gene table文件也可以。
## 输出指定路径,运行后一般输出genefamilies,pathcoverage,pathabundance三个文件和比对过程中的中间文件(包括metaphlan的结果)
humann --input $SAMPLE --output $OUTPUT_DIR合并文件
如果对多个测序文件进行分析,可以将相同的结果进行合并,再统一进行处理和分析。humann提供了很多小功能,大部分都可以对单个文件或合并文件进行处理。当然,先进行其他处理,再进行合并也是可以的,但合并文件类型必须一致。将所有需要合并且类型格式一致的文件放到同一目录下,就可以进行合并了。也可以将不同类型的文件,但不同类型使用特定名称分开,合并时使用--file_name指定文件名特定名称分别进行合并。
humann2_join_tables --input $OUTPUT_DIR --output humann2_genefamilies.tsv --file_name genefamilies_relab详细参数:
usage: humann_join_tables [-h] [-v] -i INPUT -o OUTPUT [--file_name FILE_NAME] [-s]
Join gene, pathway, or taxonomy tables
optional arguments:
-h, --help show this help message and exit
-v, --verbose additional output is printed
-i INPUT, --input INPUT
the directory of tables
-o OUTPUT, --output OUTPUT
the table to write
--file_name FILE_NAME
only join tables with this string included in the file name
-s, --search-subdirectories
search sub-directories of input folder for files功能注释
运行完成后一般得到MetaCyc通路结果,但有时候还想获得其他数据库的注释结果,humann提供转换的功能结果有rxn, go, ko, level4ec, pfam, eggnog,使用genefamilies文件mapping到对应的数据库,--groups指定数据库,获得功能注释结果。
humann_regroup_table --input $TABLE --groups $GROUPS --output $TABLE2详细参数
usage: humann_regroup_table [-h] [-i INPUT] [-g {uniref90_rxn,uniref50_rxn,uniref50_go,uniref90_go,uniref50_ko,uniref90_ko,uniref50_level4ec,uniref90_level4ec,uniref50_pfam,uniref90_pfam,uniref50_eggnog,uniref90_eggnog}] [-c CUSTOM]
[-r] [-f {sum,mean}] [-e PRECISION] [-u {Y,N}] [-p {Y,N}] [-o OUTPUT]
HUMAnN utility for regrouping table features
=============================================
Given a table of feature values and a mapping
of groups to component features, produce a
new table with group values in place of
feature values.
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
Original output table (tsv or biom format); default=[TSV/STDIN]
-g {uniref90_rxn,uniref50_rxn,uniref50_go,uniref90_go,uniref50_ko,uniref90_ko,uniref50_level4ec,uniref90_level4ec,uniref50_pfam,uniref90_pfam,uniref50_eggnog,uniref90_eggnog}, --groups {uniref90_rxn,uniref50_rxn,uniref50_go,uniref90_go,uniref50_ko,uniref90_ko,uniref50_level4ec,uniref90_level4ec,uniref50_pfam,uniref90_pfam,uniref50_eggnog,uniref90_eggnog}
Built-in grouping options
-c CUSTOM, --custom CUSTOM
Custom groups file (.tsv or .tsv.gz format)
-r, --reversed Custom groups file is reversed: mapping from features to groups
-f {sum,mean}, --function {sum,mean}
How to combine grouped features; default=sum
-e PRECISION, --precision PRECISION
Decimal places to round to after applying function; default=3
-u {Y,N}, --ungrouped {Y,N}
Include an 'UNGROUPED' group to capture features that did not belong to other groups? default=Y
-p {Y,N}, --protected {Y,N}
Carry through protected features, such as 'UNMAPPED'? default=Y
-o OUTPUT, --output OUTPUT
Path for modified output table; default=STDOUT标准化/归一化
输出结果中,genefamilies是以RPK(reads per kilobase)形式输出的。使用genefamilies文件获得的功能注释结果也是RPK结果,如果想对数据进行归一化处理,可以使用humann_renorm_table来处理,输出形式有两种,copies per million [cpm], relative abundance [relab],一般默认都是相对丰度。另外还要考虑是否去掉UNMAPPED, UNINTEGRATED, UNGROUPED这三种结果。毕竟这三种结果占比很大。
humann_renorm_table --input $SAMPLE_genefamilies.tsv --output $SAMPLE_genefamilies_relab.tsv --units relab --special n详细参数:
usage: humann_renorm_table [-h] [-i INPUT] [-u {cpm,relab}] [-m {community,levelwise}] [-s {y,n}] [-p] [-o OUTPUT]
HUMAnN utility for renormalizing TSV files
===========================================
Each level of a stratified table will be
normalized using the desired scheme.
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
Original output table (tsv or biom format); default=[TSV/STDIN]
-u {cpm,relab}, --units {cpm,relab}
Normalization scheme: copies per million [cpm], relative abundance [relab]; default=[cpm]
-m {community,levelwise}, --mode {community,levelwise}
Normalize all levels by [community] total or [levelwise] totals; default=[community]
-s {y,n}, --special {y,n}
Include the special features UNMAPPED, UNINTEGRATED, and UNGROUPED; default=[y]
-p, --update-snames Update '-RPK' in sample names to appropriate suffix; default=off
-o OUTPUT, --output OUTPUT
Path for modified output table; default=[STDOUT]重命名
对特征进行重命名,跟regroup不同,重命名是根据mapping数据库对genefamilies进行指定命名,-n指定重命名结果(kegg-orthology,kegg-pathway,kegg-module,ec,metacyc-rxn,metacyc-pwy,pfam,eggnog,go,infogo1000)。区别regroup是,只对feature重命名,不进行合并计算。
humann_rename_table --input $TABLE --names $NAMES --output $TABLE2详细参数:
usage: humann_rename_table [-h] [-i INPUT] [-n {kegg-orthology,kegg-pathway,kegg-module,ec,metacyc-rxn,metacyc-pwy,pfam,eggnog,go,infogo1000}] [-c CUSTOM] [-s] [-o OUTPUT]
HUMAnN utility for renaming table features
===========================================
For additional name mapping files, run the following command:
$ humann_databases --download utility_mapping full $DIR
Replacing, $DIR with the directory to download and install the databases.
optional arguments:
-h, --help show this help message and exit
-i INPUT, --input INPUT
Original output table (tsv or biom format); default=[TSV/STDIN]
-n {kegg-orthology,kegg-pathway,kegg-module,ec,metacyc-rxn,metacyc-pwy,pfam,eggnog,go,infogo1000}, --names {kegg-orthology,kegg-pathway,kegg-module,ec,metacyc-rxn,metacyc-pwy,pfam,eggnog,go,infogo1000}
Table features that can be renamed with included data files
-c CUSTOM, --custom CUSTOM
Custom mapping of feature IDs to full names (.tsv or .tsv.gz)
-s, --simplify Remove non-alphanumeric characters from names
-o OUTPUT, --output OUTPUT
Path for modified output table; default=[STDOUT]其他功能
上述列出的功能是较为常用的功能,humann还包含其他功能,具体可以通过help或者humann user manual查看。
humann_barplot
humann_strain_profiler
humann_benchmark
humann_genefamilies_genus_level
humann_reduce_table
humann_rna_dna_norm
humann_build_custom_database
humann_humann1_kegg
humann_regroup_table
humann_split_stratified_table
humann_unpack_pathways
humann_associate
humann_infer_taxonomy
humann_split_table附录-参数详情
usage: humann [-h] [--version] [-v] [-r] [--bypass-prescreen] [--bypass-nucleotide-index] [--bypass-translated-search]
[--bypass-nucleotide-search] -i <input.fastq> -o <output> [--nucleotide-database <nucleotide_database>]
[--annotation-gene-index <3>] [--protein-database <protein_database>] [--evalue <1.0>]
[--search-mode {uniref50,uniref90}] [--metaphlan <metaphlan>] [--metaphlan-options <metaphlan_options>]
[--diamond-options <diamond_options>] [--bowtie-options <bowtie_options>] [--o-log <sample.log>]
[--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}] [--remove-temp-output] [--threads <1>]
[--prescreen-threshold <0.01>] [--nucleotide-identity-threshold <0.0>]
[--translated-identity-threshold <Automatically: 50.0 or 80.0, Custom: 0.0-100.0>]
[--translated-subject-coverage-threshold <50.0>] [--nucleotide-subject-coverage-threshold <50.0>]
[--translated-query-coverage-threshold <90.0>] [--nucleotide-query-coverage-threshold <90.0>]
[--bowtie2 <bowtie2>] [--usearch <usearch>] [--rapsearch <rapsearch>] [--diamond <diamond>]
[--taxonomic-profile <taxonomic_profile.tsv>] [--id-mapping <id_mapping.tsv>]
[--translated-alignment {usearch,rapsearch,diamond}] [--xipe {on,off}] [--minpath {on,off}]
[--pick-frames {on,off}] [--gap-fill {on,off}] [--output-format {tsv,biom}] [--output-max-decimals <10>]
[--output-basename <sample_name>] [--remove-stratified-output] [--remove-column-description-output]
[--input-format {fastq,fastq.gz,fasta,fasta.gz,sam,bam,blastm8,genetable,biom}]
[--pathways-database <pathways_database.tsv>] [--pathways {metacyc,unipathway}]
[--memory-use {minimum,maximum}]
HUMAnN : HMP Unified Metabolic Analysis Network 2
optional arguments:
-h, --help show this help message and exit
--version show program's version number and exit
-v, --verbose additional output is printed
-r, --resume bypass commands if the output files exist
--bypass-prescreen bypass the prescreen step and run on the full ChocoPhlAn database
--bypass-nucleotide-index
bypass the nucleotide index step and run on the indexed ChocoPhlAn database
--bypass-translated-search
bypass the translated search step
--bypass-nucleotide-search
bypass the nucleotide search steps
-i <input.fastq>, --input <input.fastq>
input file of type {fastq,fastq.gz,fasta,fasta.gz,sam,bam,blastm8,genetable,biom}
[REQUIRED]
-o <output>, --output <output>
directory to write output files
[REQUIRED]
--nucleotide-database <nucleotide_database>
directory containing the nucleotide database
[DEFAULT: /home/dengysh/anaconda3/envs/mpa/humann_database/chocophlan_v296_201901]
--annotation-gene-index <3>
the index of the gene in the sequence annotation
[DEFAULT: 3]
--protein-database <protein_database>
directory containing the protein database
[DEFAULT: /home/dengysh/anaconda3/envs/mpa/humann_database/uniref90_v201901]
--evalue <1.0> the evalue threshold to use with the translated search
[DEFAULT: 1.0]
--search-mode {uniref50,uniref90}
search for uniref50 or uniref90 gene families
[DEFAULT: based on translated database selected]
--metaphlan <metaphlan>
directory containing the MetaPhlAn software
[DEFAULT: $PATH]
--metaphlan-options <metaphlan_options>
options to be provided to the MetaPhlAn software
[DEFAULT: "-t rel_ab"]
--diamond-options <diamond_options>
options to be provided to the diamond software
[DEFAULT: "--top 1 --outfmt 6"]
--bowtie-options <bowtie_options>
options to be provided to the bowtie software
[DEFAULT: "--very-sensitive"]
--o-log <sample.log> log file
[DEFAULT: temp/sample.log]
--log-level {DEBUG,INFO,WARNING,ERROR,CRITICAL}
level of messages to display in log
[DEFAULT: DEBUG]
--remove-temp-output remove temp output files
[DEFAULT: temp files are not removed]
--threads <1> number of threads/processes
[DEFAULT: 1]
--prescreen-threshold <0.01>
minimum percentage of reads matching a species
[DEFAULT: 0.01]
--nucleotide-identity-threshold <0.0>
identity threshold for nuclotide alignments
[DEFAULT: 0.0]
--translated-identity-threshold <Automatically: 50.0 or 80.0, Custom: 0.0-100.0>
identity threshold for translated alignments
[DEFAULT: Tuned automatically (based on uniref mode) unless a custom value is specified]
--translated-subject-coverage-threshold <50.0>
subject coverage threshold for translated alignments
[DEFAULT: 50.0]
--nucleotide-subject-coverage-threshold <50.0>
subject coverage threshold for nucleotide alignments
[DEFAULT: 50.0]
--translated-query-coverage-threshold <90.0>
query coverage threshold for translated alignments
[DEFAULT: 90.0]
--nucleotide-query-coverage-threshold <90.0>
query coverage threshold for nucleotide alignments
[DEFAULT: 90.0]
--bowtie2 <bowtie2> directory containing the bowtie2 executable
[DEFAULT: $PATH]
--usearch <usearch> directory containing the usearch executable
[DEFAULT: $PATH]
--rapsearch <rapsearch>
directory containing the rapsearch executable
[DEFAULT: $PATH]
--diamond <diamond> directory containing the diamond executable
[DEFAULT: $PATH]
--taxonomic-profile <taxonomic_profile.tsv>
a taxonomic profile (the output file created by metaphlan)
[DEFAULT: file will be created]
--id-mapping <id_mapping.tsv>
id mapping file for alignments
[DEFAULT: alignment reference used]
--translated-alignment {usearch,rapsearch,diamond}
software to use for translated alignment
[DEFAULT: diamond]
--xipe {on,off} turn on/off the xipe computation
[DEFAULT: off]
--minpath {on,off} turn on/off the minpath computation
[DEFAULT: on]
--pick-frames {on,off}
turn on/off the pick_frames computation
[DEFAULT: off]
--gap-fill {on,off} turn on/off the gap fill computation
[DEFAULT: on]
--output-format {tsv,biom}
the format of the output files
[DEFAULT: tsv]
--output-max-decimals <10>
the number of decimals to output
[DEFAULT: 10]
--output-basename <sample_name>
the basename for the output files
[DEFAULT: input file basename]
--remove-stratified-output
remove stratification from output
[DEFAULT: output is stratified]
--remove-column-description-output
remove the description in the output column
[DEFAULT: output column includes description]
--input-format {fastq,fastq.gz,fasta,fasta.gz,sam,bam,blastm8,genetable,biom}
the format of the input file
[DEFAULT: format identified by software]
--pathways-database <pathways_database.tsv>
mapping file (or files, at most two in a comma-delimited list) to use for pathway computations
[DEFAULT: metacyc database ]
--pathways {metacyc,unipathway}
the database to use for pathway computations
[DEFAULT: metacyc]
--memory-use {minimum,maximum}
the amount of memory to use
[DEFAULT: minimum]参考
https://huttenhower.sph.harvard.edu/humann3/ https://github.com/biobakery/humann Nature子刊:HUMAnN2实现宏基因组和宏转录组种水平功能组成分析