病原微生物扩增子数据分析实战(二):fastp软件进行质量控制

接上一篇,数据拆分完成后,得到 FASTQ 文件,下面对数据进行质控。
当前主流测序平台的数据存储格式无外乎两种,FASTQ(Illumina, MGI),BAM(Life Ion Torrent,PacBio),对于 BAM 文件,通常也需要先转换成 FASTQ 文件后再进行质控处理。
质控软件非常多,有 FastQC,Cutadapt, Trimomatic 等,通常需要多款软件共同配合使用,这难免过于繁琐,在实际项目中,推荐用fastp,根据官网介绍,这是一款处理 FASTQ 文件的 all-in-one 软件,质控用它就够了,下面是扩增子数据的质控命令。
/path/to/fastp -i $fq1 -I $fq2 \
-h ${outdir}/${sample}_merge_clean.html \
-j ${outdir}/${sample}_merge_clean.json \
-m --merged_out ${outdir}/${sample}_merge_clean.fastq.gz \
--failed_out ${outdir}/${sample}_failed.fastq.gz \
--include_unmerged \
--overlap_len_require 6 \
--overlap_diff_percent_limit 20 \
--detect_adapter_for_pe \
-5 \
-r \
-l 20 \
-n 5\
-y \
--thead 10
参数解释:
-i $fq1,输入样本的 FASTQ1 文件,可以是 gz 压缩格式;
-I $fq2,输入样本的 FASTQ2 文件,可以是 gz 压缩格式;
-h ${outdir}/${sample}_merge_clean.html,输出 html 格式的质控报告;
-j ${outdir}/${sample}_merge_clean.json,输出 json 格式的质控报告;
-m,合并双端 reads 模式,设定该参数时满足条件的双端 reads 会合并在一起;
--merged_out ${outdir}/${sample}_merge_clean.fastq.gz,输出合并后的结果文件;
--failed_out ${outdir}/${sample}_failed.fastq.gz,保存被过滤掉的 reads 到该文件中;
--include_unmerged,设定该参数,未能合并的 reads 也包含在结果文件中,否则默认是不包含的;
--overlap_len_require 6,合并的条件一:双端 reads 至少有 6bp 重叠;
--overlap_diff_percent_limit 20,合并条件二:重叠区域最多允许有 20%的碱基错配;
--detect_adapter_for_pe,自动检测双端测序的接头序列并切除,默认只自动检测单端数据的接头序列;
-5,从 5'端开始滑动一个窗口,如果窗口内碱基的平均质量低于某个阈值,则剪切掉窗口内的序列,否则停止剪切;
-r, 从 5'端开始滑动一个窗口,如果窗口内碱基的平均质量低于某个阈值,则剪切掉窗口内以及其后的所有序列;
-l 20,丢弃长度低于 20bp 的序列;
-n 5,read 中 N 碱基数不能超过 5 个;
-y,低复杂度过滤;
--thead 10,指定 10 条工作线程;
以上命令除了常规的剪切、过滤操作外,还进行了双端 reads 的合并,最终结果如下 :
wenku1
|___wenku1_failed.fastq.gz 被过滤掉的reads结果文件
|___wenku1_merge_clean.fastq.gz 通过质控的reads结果文件
|___wenku1_merge_clean.html html质控报告
|___wenku1_merge_clean.json json质控报告
通过 html 质控报告,可以直观地看出很多指标,如过滤前后的 reads 数、bases 数、碱基质量、插入片段长度、碱基组成、GC 含量、接头类别等,如:


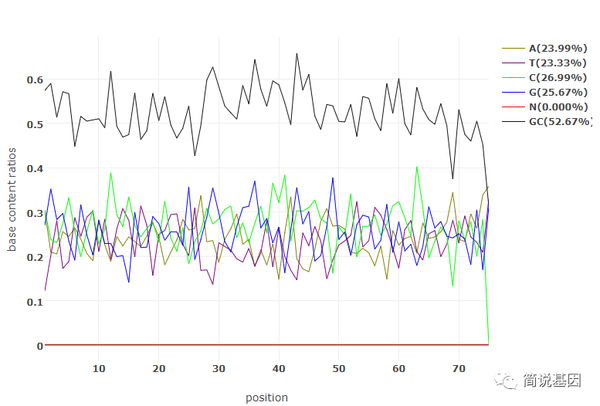

此外,也可以通过编写脚本读取 json 报告的内容,生成个性化的质控报告。
质控知识小结
数据质控的目的是为了过滤掉那些不好的序列,从而减小对下游数据分析的干扰。
下面是一条 FASTQ 格式的序列,我们能对它进行些什么操作呢?主要有剪切、过滤、碱基质量校正等。
@M06862:6:000000000-C9TL7:1:1102:17871:1939 1:N:0:ATTACTCG+GGCTCTGA
GCCCTCTTTGTCCTCCTTGGTGAGAACCAGGGCCTCGACCTCATCGCCCACGGAAACGACCTCGTTGGGGTCGA
+
1111>>CFF3F@EGEEGGB1G1BGHGHHGFA0AAFF0EEGGHGHHBEEE?FGCEGGHFCEEGHAHGHFC?GG??
为什么要剪切 read,无非是因为 read 中要么含有接头,要么含有低质量序列,这些信息对我们没用,甚至对分析结果造成干扰。
1.接头序列剪切
如果 read 中含有接头序列,则从接头开始的地方删除至 read 末尾。接头序列的来源可能是因为测序片段的长度低于测序的读长,从而被测通导致的,另外也不排除接头之间形成了二聚体,测出来的只有接头序列。
2.低质量序列剪切
Illumina 测序仪的特性,低质量序列可能位于 5'端,3'端或者 read 的中间,对应的处理方式有这几种:
- 从 5'端开始滑动一个窗口,如果窗口内碱基的平均质量低于某个阈值,则剪切掉窗口内的序列,否则停止剪切
- 从 3'端开始滑动一个窗口,如果窗口内碱基的平均质量低于某个阈值,则剪切掉窗口内的序列,否则停止剪切
- 从 5'端开始滑动一个窗口,如果窗口内碱基的平均质量低于某个阈值,则剪切掉窗口内以及其后的所有序列
通过以上几种方法,就可以剪切掉 5'端和 3'端的低质量序列,如果低质量序列位于 read 中间,则剪切掉该低质量序列及其后的所有序列。
3.全局剪切
以上去掉低质量序列的方式是有条件的,需要满足一定的阈值才会进行,也可以直接剪切掉 read 的头部和尾部,通常做法是:
- 去掉 5'端一定数量碱基
- 去掉 3'端一定数据碱基
- 或者限定 read 的最大长度,当 read 的长度超过限定值时,其尾部序列会被剪切掉
4.polyG 剪切
双色发光法的 Illumina 设备(NextSeq /NovaSeq),在没有光信号情况下 base calling 的结果会返回 G,所以在序列的尾端可能会出现较多的 polyG,需要被去除。
5.polyX
如果 3'端存在 polyX(如 mRNA-Seq 数据中的 polyA),可以剪切掉。
完成了剪切,下面就是过滤了。
1.质量过滤
对于低质量 reads,应直接丢弃,有如下方式:
- 按低质量碱基占 read 的比例,如达到 40%,则过滤掉,当然需要先定义低质量碱基的阈值,如 phred quality < Q15
- 按 read 中碱基的平均质量,如低于 30,则过滤掉
2.N 碱基过滤
测序过程中某个碱基无法识别时,体现在 read 中可能是一个大写 N 字母,当这样的 N 碱基过多时,则过滤掉该 read。
3.低复杂度过滤
复杂度的定义是 read 中与下一个碱基不同的碱基的百分比(base[i] != base[i+1])。
# a 51-bp sequence, with 3 bases that is different from its next base
seq ='AAAATTTTTTTTTTTTTTTTTTTTTGGGGGGGGGGGGGGGGGGGGGGCCCC'
complexity = 3/(51-1) = 6%
这样的序列在靶向测序中,通常是不应该存在的,因此需要去除。
4.长度过滤
过滤掉太短或太长的序列:
- 长度太短,过滤掉
- 长度太长,过滤掉
此外,为了降低测序错误产生的噪声,质控时还可以对碱基的质量进行校正,通常的做法有:
1.PE 数据碱基校正
当双端测序的配对 reads 之间有 overlap 并且有错配时,如果错配碱基一个质量很高,一个质量很低,则进行碱基及其质量的校正。
2.UMI 标签
通过 UMI 标签来消除重复和测序错误,这通常在超高深度的测序数据中用到。
至此,质控部分就讲完了,下一篇介绍聚类分析。
上一篇回顾:
