
如何画热图

前言
这篇文章的标题非常low,因为热图是生信中最为常见、普遍的图形可视化方式,大多用以展现某种特定的基因表达分布模式,而绘制热图相信是每一个了解生信的人,耳熟能详的必备技能。
今天这篇文章,不会涉及到“用哪个软件去画热图呀”“应该调哪个包”“距离函数与linkage函数什么意义?怎么选?如何实现?”“什么时候该做row normalization”这类术的问题,因为相信善用搜索引擎的话,可以找到不少这类教程。今天谈的问题,我希望是一个道的问题:如何绘制一幅真正有用、能放到paper上的热图?可能的话,我尽可能把它拔高到一个流程设计的角度来俯瞰这个问题,希望能真正的终结一类单因子多组或简单双因子多组实验设计的生物学表达谱分析问题。
既然是讨论画“有用”的热图,那么一开始就要排斥掉一些“没用”的热图,“没用”的热图,我把它分为两类:

- “脱裤子放屁然并卵”热图。就是上面这幅非常亮瞎眼这种,我相信绝大部分热图可以归到这类。实验设计非常简单,2组,3 vs 3,差异表达基因的定义很明了,不是上调就是下调,通过阈值筛选以后,图形展示结果是早可以预见的,毫无意外。既然这样,为什么不直接列个表,况且根本没有GeneSymbol,无法做下游解读。所以,综上,这就是一个“田”字的色盲测试图。
- “功夫在戏外”热图。统计学家会这么建议使用热图。热图是一个非常有价值的“探索性分析”工具,可以配合非监督的聚类来观察各种有趣的数据性质,提供进行下游分析处理时的决断与线索,比如Spike-in的表达模式?持家基因的表达模式?不同方差区段的基因表达模式?哪几个样本有问题?是不是有Batch Effect要校正?怎么校用什么校?但对于一篇正常的生物学研究文章来说,这种就属于“锦衣夜行”,上不得台面,所以这里也略过。
示例数据来源与实验设计
为了言之有物,方便演示与说明,我们需要一个数据集,这里就挑了GSE18290中人胚胎不同发育时期表达数据。选这个数据原因在于相对各种敲降、加药、抑制的实验组合,这个数据集不需要太多背景了解,实验设计也很简单,一目了然:


实验目的也很明了,通过比较不同发育时期胚胎的表达谱差异,找出是哪些Pathway,生物学功能发生了改变。
今天写这篇文章的目的,其实是为了与上一篇生物功能富集检验 - 生信闲扯 - 知乎专栏做一个联动,实际演示一下,如何利用已有算法的特性,去拼接、组装出一个新的分析算法来。所以,如果现在还未读过生物功能富集检验 - 生信闲扯 - 知乎专栏的话,那么请先花时间理解一下上一篇的内容。
在开始新的分析方法的讲解前,按照传统,必须回顾下经典的分析思路,也就是各位读过的绝大多数涉及表达谱分析的文献时,遇到此类实验设计,是如何进行下游分析,然后绘出热图的。
然后我们可以比较下,各种分析思路的优劣势、长短处。
经典思路——先做聚类再检功能
上面标题里,这个“聚类”是广义的,指代所有“找出表达轮廓相近基因”的这类方法。经典分析思路来绘制热图,它的逻辑链条如下:
- 由统计或者机器学习方法,将表达轮廓与普通基因有明显差异的基因检出。这个方法,可以是各类变种的ANOVA,也可以是STEM这类专门针对时序设计的算法。这一步需要特别注意的是,在经典思路中,大规模的基因过滤发生在此处。
- 由层次聚类方法,将表达轮廓相似的基因归类。
- 绘制热图,展示表达模式,并尝试使用第一类生物功能富集检验方法来解释归类后的基因列表。使用的是第一类方法,比如Fisher 's exact test,原因在于第一步上发生了大规模过滤。
以下实际演示一下,在胚胎发育GSE18290数据集里,这个分析思路的效果。简单起见,下面只用One-way ANOVA过滤以绕开繁琐的算法说明(这里实际上再配合STEM的检出过滤会得到一个更“好看”的热图,但这个分析思路的缺陷却是不会变的)
F = \frac{\sigma^2_{\mathrm{between}}}{\sigma^2_{\mathrm{within}}}- 首先,使用 One-way ANOVA 的 F-test 显著性,将基因排序。这里ANOVA中F-test的本质,在于将基因按照 “组间差异越大而组内差异越小” 这个标准 (见上述公式),进行排序
- 设定每个基因必须至少 IQR >= 0.585,即仅保留在所有样本中,第一四分位数与第三四分位数差值至少有 1.5 倍 Fold Change 的基因。(注意这里需要设定个阈值过滤)
- 取上述满足1、2条件的前200个基因。(这里取 200 个是个经验值,主要为了后继演示的方便)
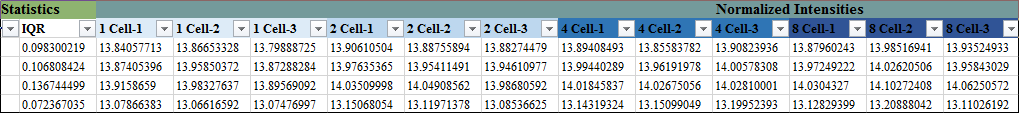
这里提一下,上述步骤2是必不可少的,因为在高通量分析里,任何涉及方差估计的普通统计检验方法都是容易出问题的,比如,第一步的基于One-way ANOVA的F排序,它的前4个基因是这样的:

经过上述三步处理,可以把相关基因拿出来做热图了。可以绘出下面这张热图:
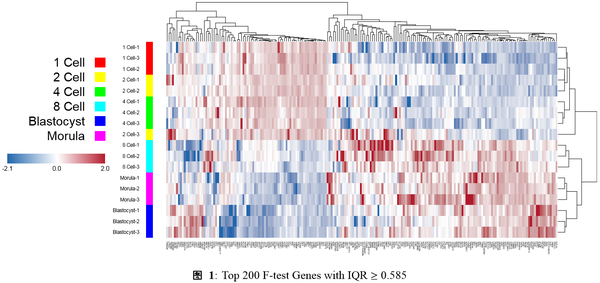
当终于绘出上述热图时,才会遇上真正的困难:如何把这幅热图,嵌入生物学文章内?如何展开讨论?它说明了一个什么生物学问题?
有了上面这幅图,再结合一点初高中生物学常识,我们还是可以掰一下的对不?比如:从热图上看,1细胞、2细胞与4细胞期,这几个时间点上表达谱变化非常相似,这可能与这几个时期的细胞都是全能干细胞有关。到了8细胞期时,胚胎开始初步出现极性,桑椹胚与囊胚期,开始出现胚层分化,全能干细胞开始分化成多能干细胞,因此这个也与热图的观察结果一致:对应三组细胞的差异表达变化开始加剧。
掰的好像挺有道理的对不?但这终究只是一个总体上的概括,在真正的生物学研究论文里,需要更为具体的描述:这个热图展示的特定的表达模式,说明了哪些生物学功能与信号通路的改变?是什么导致了这些改变?这种改变又会产生什么样的后果?能回答第一个问题的,是一篇合格的论文,能接着回答第二、第三个问题的,则是优秀的论文。
作为一个生信分析的结果,那么至少要对第一个问题的回答有帮助。这个示例里,将接着使用上面这个热图,去试着回答第一个问题,看看能做哪些尝试。一般的套路是:
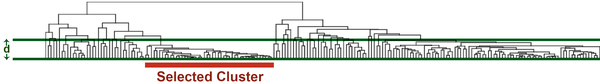

- 选择一个合适的基因间差异大小阈值d,切割聚类树
- 选中一个合适大小S的geneset
- 进行Fisher ’s exact test,将检出的富集生物学功能,标注在原先的热图上
如果是计算机出身的读者,可能这个时候,已经在拍大腿了:麻痹,这不就是个递归树形结构的深度优先遍历问题么?本科数据结构问题好么?对不对?比如,可以这么做:
- 聚类就是生成一个多叉树,叶是基因,节点是子树或叶的集合,节点本身可以存储树高d,与总叶数目s。
- 找基因簇的问题,转化成深度优先遍历子树,在合适的d和s处,剪枝求得所有叶的基因列表。
- Fisher 's exact test,Problem Solved!
好的,如果真是上面这么想,准备撸袖子管去实现的话,那么我要拍肩膀来说一句了:恭喜您,您进坑了!!!
因为从一个流程设计的视角上去看,“合适”这个词随着数据不同是很难精确定义的,上面每一个步骤的参数,只要有一个微小的扰动,都可能带来完全不同的检测结果,这个流程可能会产生一个好看的花瓶,但是一碰就碎!
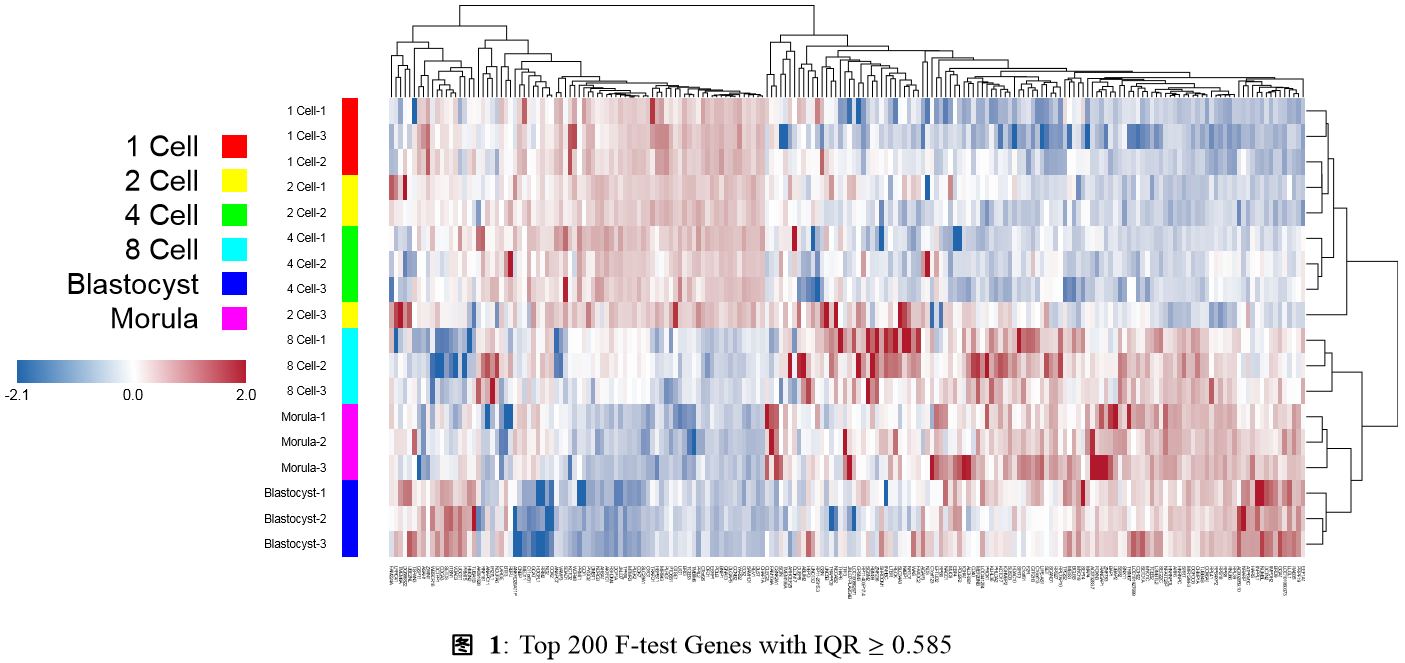
不管怎么样,我们先开一下挂,试一下,这个流程走下去的结果如何。比如我们先开启了写轮眼模式以后,挑出了一个觉得“合适”的基因簇,嘛,就是上面红色标出来的那坨,大概45个左右基因吧。然后拷贝粘贴下基因列表,找个免费、开源的检测工具弄下,比如DAVID在线分析工具 (原理:Fisher 's exact test)。整理下表格,就是下面左下那张。
呜嗷,这45基因里面,有6个左右是与DNA损伤反应有关的,然后在原先热图上,这45个基因右侧,用大字写上“Response to DNA Damage”,最后再用红字标一下:FDR < 0.1,以示严谨。


我特别用绿色的色块在上图右侧里,标注了实际命中的6个基因在簇中的分布。我们可以想一下,通过这种方法,标注的基因簇,make sense么?是Science的态度么?
这两个鳝丝,大家扪心自问过么???
新的思路——先检功能再做聚类
之前的经典思路,概括起来,那就是先找表达轮廓相似的基因,在这一步进行过滤,而后再去检归类后基因的功能。从算法上讲,这个思路很直接,但由于第一步的过滤以及之后的聚类过程是纯统计、纯数值的,所以它导致了绘图完成之后,进行生物学解读时的困难。
那么可不可以倒过来考虑:先做功能检出,再做聚类?这么做的困难在哪里?回顾一下刚才经典解决方案中的失败之处,可以有如下启发:
- 在检出功能时,需要一个稳健(受输入波动影响小)而又敏感(能检出微弱趋势)的算法,这个检出过程,需要避免大规模的基因过滤
- 在绘制热图时,仅展示有价值的基因,即过滤发生在此处
非常巧,之前的生物功能富集检验 - 生信闲扯 - 知乎专栏中,正好提到了一类功能检出算法,叫GSEA,满足上面第一点的要求。回忆一下GSEA这个算法的本质,它需要首先定义一个恰当的统计量来排序基因,其次检出具有特定生物学功能的基因,是否在一端富集。
当有了上述思考以后,问题可以被转化了:如何定义一个能够表述基因“组间差异表达程度”的统计量?在刚才的经典分析思路中,同时使用了“ANOVA F-test排序”和“IQR”过滤,绕开了这个问题。但在这里,必须正面回答这个问题,并且没有过滤发生。
之前在相应的小节里已经反复强调过了,在小样本高通量数据分析里,一定不能使用普通版本的统计检验,而一定要使用针对某种特定场景做过方差校正的特殊检验方法。这里,可以告诉大家的是,使用Moderated-F test也是不行的,它的校正力度太弱,无法完全避免微小方差基因在排序列表端部出现。
想到这一步,其实选择已经不多了。那么不卖关子了,这里其实要提取的是PAM分析的思路方法,PAM分析的全称叫做 Nearest Shrunken Centriod Classifier,然而这里并不是要做分类,而是要利用PAM score来构建一个统计量。因为PAM score的本质,就是一个Penalized/Regularized Fisher 's Linear Discriminant,用方差的经验分布来提供校正,这个是Tibshirani(Elements of Statistical Learning: data mining, inference, and prediction. 2nd Edition.作者)几个生信工具的一贯思路。
通过上面的分析,这个思路里,需要的算法、统计量的检验,将这两处任督二脉打通之后,之后的分析即可畅通无阻:
- 首先将基因按照“在不同组间差异表达”程度的大小排序
- 检出上面这个排序列表中,显著位于端部的生物学功能
- 在绘图中,仅保留每个检出功能中那些位于端部的、“确实是在组间差异表达”的基因
OK,流程有了,下面就非常简单了。可以找不同角度的生物学功能数据库、KEGG、BioCarta、GO、各种文献挖掘出来OOXX geneset、肿瘤疾病模组等等等,每个角度可以做一次检验,然后把合适的热图拼起来?很简单对不对?
下面就展示下最简单的、Broad Institute总结的50个生物学Hallmark通路上,GSE18290这个人胚胎发育数据集上的检出图形展示结果,注意哈,这个图有一个非常王八之气的Title:
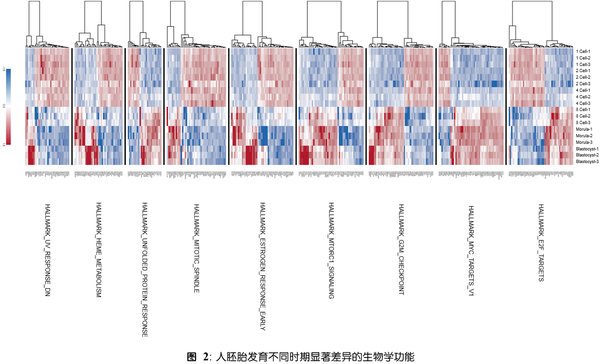

生物信息在整个的生物学研究中能起到的,往往仅仅只是指明方向、检出趋势这部分最容易的工作。拿到这份结果,对结果进行解读,不是生信专家能做到的,那是真正的生物学领域专家的战场,他们需要在各个检出的通路中找出真正关键的节点去设计下游的loss of function与gain of function实验,产生与验证假说。
比较与总结
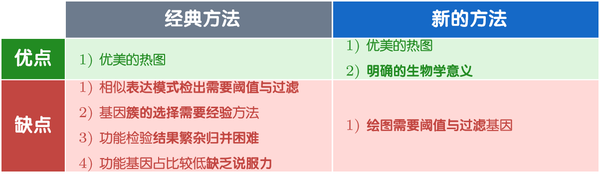
现在回顾下上两节中提到的两种思路完全相反的分析方法,总结下,它们的优缺点如下:

以上,今天的鳝丝时间到此结束。
以下为安利内容,可能引起部分人士不适。
======================================================
以下内容为广告
======================================================
看完上面的讨论,是不是跃跃欲试,想try下自己的数据?——已经没有必要自行动手了,上面的所有展示数据示例,都是自动完成的,图一是基本分析免费内容,图二需要额外收一点点辛苦费,大约就相当于招一个中等水平生信研究生鼓捣出结果需要花的补贴费吧。
请关注、垂询、充值、付费转账,然后妥妥儿的~


