


SVIM (pronounced swim) is a structural variant caller for third-generation sequencing reads. It is able to detect and classify the following six classes of structural variation: deletions, insertions, inversions, tandem duplications, interspersed duplications and translocations (see the figure below). SVIM also estimates the genotypes of deletions, insertions, inversions and interspersed duplications. Unlike other methods, SVIM integrates information from across the genome to precisely distinguish similar events, such as tandem and interspersed duplications and simple insertions. In our experiments on simulated data and real datasets from PacBio and Nanopore sequencing machines, SVIM reached consistently better results than competing methods.
Note! To analyze haploid or diploid genome assemblies or contigs, please use our other method SVIM-asm.
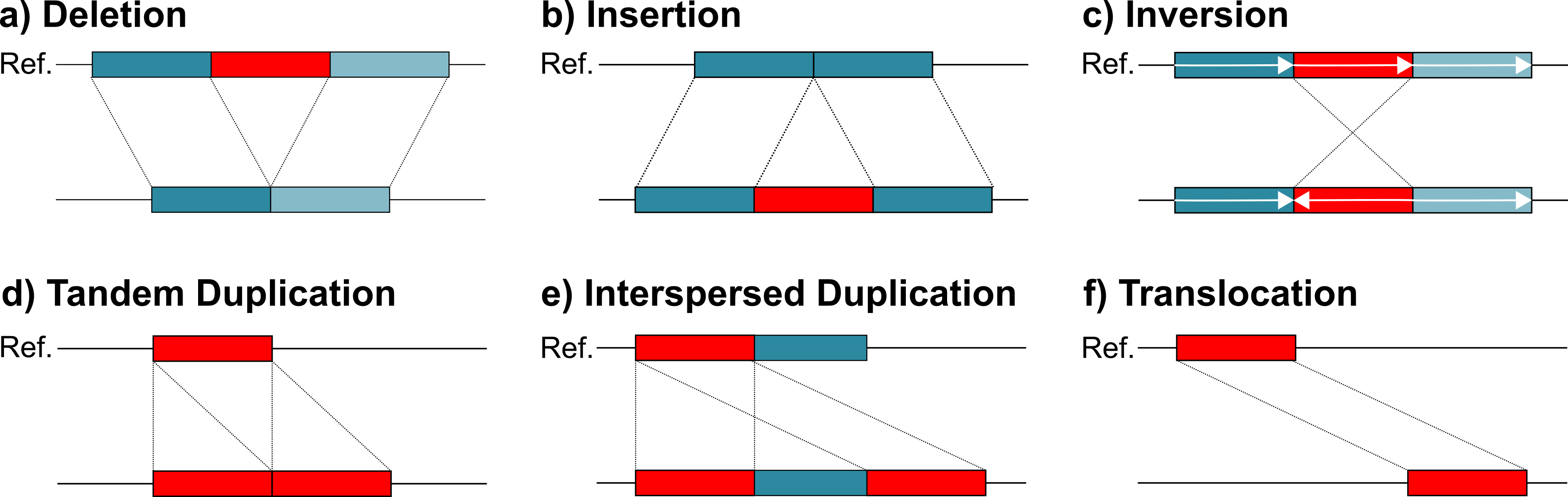
Structural variants (SVs) are typically defined as genomic variants larger than 50bps (e.g. deletions, duplications, inversions). Studies have shown that they affect more bases in an average genome than SNPs or small Indels. Consequently, they have a large impact on genes and regulatory regions. This is reflected in the large number of genetic disorders and other disease that are associated to SVs.
Next-generation sequencing technologies by providers such as Illumina generate short reads with high accuracy. However, they exhibit weaknesses in repetitive and low-complexity regions where SVs are particularly common. Single molecule long-read sequencing technologies from Pacific Biotechnologies and Oxford Nanopore produce reads with error rates of up to 15% but with lengths of several kbps. The high read lengths enable them to cover entire repeats and SVs which facilitates SV detection.
#Install via conda into a new environment (recommended): installs all dependencies including read alignment dependencies
conda create -n svim_env --channel bioconda svim
#Install via conda into existing (active) environment: installs all dependencies including read alignment dependencies
conda install --channel bioconda svim
#Install via pip (requires Python 3.6.* or newer): installs all dependencies except those necessary for read alignment (ngmlr, minimap2, samtools)
pip install svim
#Install from github (requires Python 3.6.* or newer): installs all dependencies except those necessary for read alignment (ngmlr, minimap2, samtools)
git clone https://github.com/eldariont/svim.git
cd svim
pip install .- edlib for edit distance computation
- matplotlib>=3.3.0 for plotting
- numpy and scipy for hierarchical clustering
- pysam (>=0.15.2) for SAM/BAM file processing
- pyspoa (>=0.0.6) for consensus sequence computation
- py-cpuinfo (>=7.0.0) for CPU info retrieval (checking for SIMD capabilities)
- no genotyping of tandem duplications
- genotyping assumes a diploid organism
- no built-in multi-sample variant calling
- no support for multi-threading
- no force-calling using an existing input VCF
SVIM analyzes (sorted and indexed) alignment files in BAM format. Alternatively, SVIM accepts long reads in FASTA/FASTQ format (uncompressed or gzipped) or as a file list. SVIM has been successfully tested on PacBio CLR, PacBio HiFi (CCS) and Oxford Nanopore data. It has been tested on alignment files produced by the read aligners minimap2, pbmm2 and NGMLR.
SVIM produces SV calls in the Variant Call Format (VCF). The output file variants.vcf is placed into the given working directory.
For detailed information on the usage of SVIM please see our wiki.
- v2.0.0: adds consensus sequence computation for insertions, improves clustering step (considers sequence similarity when clustering insertions and prevents signatures from same read to be clustered together), outputs sequence alleles for all SV types except BNDs and DUPs by default, updates default parameters, bugfixes
- v1.4.2: fixes invalid start coordinates in VCF output, issues warning for invalid characters in contig names
- v1.4.1: improves clustering of translocation breakpoints (BNDs), improves --all_bnds mode, bugfixes
- v1.4.0: fixes and improves clustering of insertions, adds option --all_bnds to output all SV classes in breakend notation, updates default value of --partition_max_distance to avoid very large partitions, bugfixes
- v1.3.1: small changes to partitioning and clustering algorithm, adds two new command-line options to output duplications as INS records in VCF, removes limit on number of supplementary alignments, removes q5 filter, bugfixes
- v1.3.0: improves BND detection, adds INFO:ZMWS tag with number of supporting PacBio wells, adds sequence alleles for INS, adds FORMAT:CN tag for tandem duplications, bugfixes
- v1.2.0: adds 3 more VCF output options: output sequence instead of symbolic alleles in VCF, output names of supporting reads, output insertion sequences of supporting reads
- v1.1.0: outputs BNDs in VCF, detects large tandem duplications, allows skipping genotyping, makes VCF output more flexible, adds genotype scatter plot
- v1.0.0: adds genotyping of deletions, inversions, insertions and interspersed duplications, produces plots of SV length distribution, improves help descriptions
- v0.5.0: replaces graph-based clustering with hierarchical clustering, modifies scoring function, improves partitioning prior to clustering, improves calling from coordinate-sorted SAM/BAM files, improves VCF output
- v0.4.4: includes exception message into log files, bug fixes, adds tests and sets up Travis
- v0.4.3: adds support for coordinate-sorted SAM/BAM files, improves VCF output and increases compatibility with IGV and truvari, bug fixes
If you experience any problems or have suggestions please create an issue or a pull request.
Feel free to read and cite our paper in Bioinformatics: https://doi.org/10.1093/bioinformatics/btz041. Please note that since its publication in 2019 some parts of SVIM were modified (e.g. the clustering method) while others were added (e.g. the genotyping feature).
The project is licensed under the GNU General Public License.