How to map cell fate to branches? #219
Comments
|
Hi @ClarkGregg sorry for the delay in response. I will answer your questions in the following: |
|
Hello, Following your explanation of BEAM
So pre-branch in case of testing node 3 should rather be the state 1 branch as it is leading towards pseudotime 0 ? |
|
Hello, |




|
Hi@Xiaojieqiu. Sincere thanks for your explanation. May I ask why the pre-branch will only include cells from state 2 when testing for the difference between state4 and state7. Thanks!
|
|
I have same question. Why only state 2 is the pre-branch? |
Dear all.
I am working on single cell RNA-seq data using your R package: Monocle. While I am performing pseudotime analysis, I have some questions, hopefully you can clear my mind.
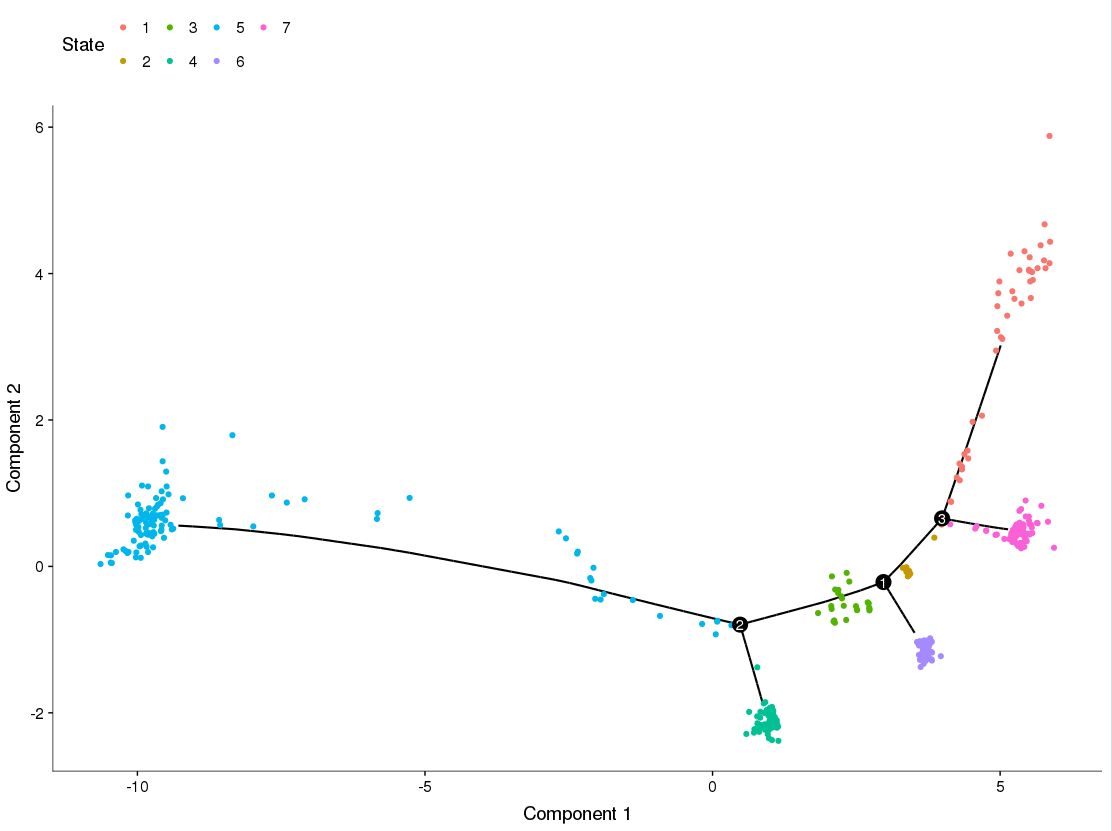
By following the pipeline posted on the website, I constructed a trajectory with several branches (States). Then I sought to perform differential expression test to identify genes expressed in a branch-dependent manner. Here are my question one: I used BEAM() function inside Monocle to do it, I tried to compare state 1 and state 7, or equally branch point 3 and visualize my result using plot_genes_branched_heatmap() function. I am wondering, if the 'pre-branch' state is comprised of all states excepted for the two being tested or just state 2, in this particular case. And also what is the 'pre-branch' if I try to compare states that sit within different branches, say, branch 4 and branch 7.
My question 2 is about a parameter, the parameter inside plot_genes_branched_heatmap() called branch_label. the default branch label is 'cell fate 1' and 'cell fate 2'. How do I map these labels to the corresponding branches being tested? For example, if I am comparing branch 1 and branch 7. should 'cell fate 1' correspond to branch 1 and branch 7 correspond to 'cell fate 2' ?
The attachment is the trajectory I constructed.
Thank you in advance
The text was updated successfully, but these errors were encountered: