Requirements & Usage
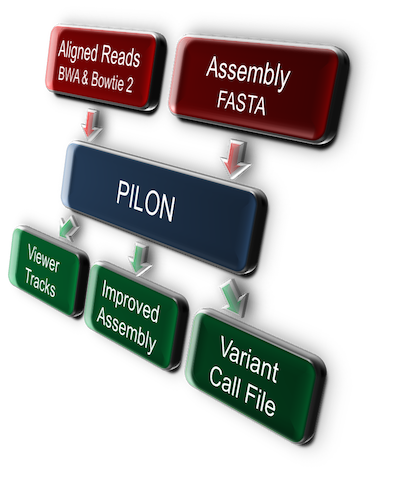
Pilon is distributed as a single jar file. Requirements to run it include:
- Java runtime 1.7 or later
- 8GB or more memory to allocate to the JVM. The amount of memory required depends on the genome, the read data, and how many fixes Pilon needs to make. Generally, bacterial genomes with ~200x of Illumina coverage will require at least 8GB, though 16GB is recommended.
- Larger genomes will require more memory to process; exactly how much is very data-dependent, but as a rule of thumb, try to allocate 1GB per megabase of input genome to be processed.
Input to Pilon consists of the input genome in FASTA format along with one or more BAM files of reads aligned to the input genome.
Pilon requires a FASTA of the input genome, either a draft assembly or a reference to be used for variant calling. If the input is an assembly to be improved, each scaffold should be a FASTA element, with at least 10 consecutive Ns representing gaps between contigs within the scaffold.
Pilon also requires one or more BAM files of reads aligned to the input genome. Pilon can use three types of BAM files:
--frags <frags.bam>
for paired-end sequencing of DNA fragments, such as Illumina paired-end reads of fragment size <1000bp.
--jumps <jumps.bam>
for paired sequencing data of larger insert size, such as Illumina mate pair libraries, typically of insert size >1000bp.
--unpaired <unpaired.bam>
for unpaired sequencing reads.
Starting with Pilon version 1.17, Pilon can be given any type of BAM file with the command line argument --bam <any.bam>, and it will automatically classify the BAM as one of the three types above.
Input BAM files must be sorted in coordinate order and indexed. You may get the following message if your BAM index file is missing or older than the BAM file itself:
WARNING: BAM index file "path/to/bai_file" is older than BAM "/path/to/bam_file".
To resolve this issue run:
samtools index /path/to_bam_file
Pilon makes considerable use of pairing information where available, and Pilon assumes the aligner is able to set the “proper pair” flag correctly based on the separation and orientation of the reads in the pair. Prior to Pilon version 1.11, Pilon also assumed that the pairs are in Forward-Reverse (FR) orientation in the BAM files. Note that standard Illumina paired jumping libraries are sequenced in RF orientation. This means that for those early versions of Pilon, Illumina jump libraries need to be flipped prior to alignment. With version 1.11 and later, Pilon will determine the orientation of the mate pairs automatically, so flipping jumps to FR prior to alignment is not necessary.
Pilon development was done primarily using BAM files produced by the aligners BWA (using sampe or mem for paired alignment) and Bowtie 2, but other conforming aligners should also work.
Pilon's command line options are described below, which is also the output from the --help argument. The pilon command below is shorthand for the Java Virtual Machine (JVM) invocation, which is usually something like:
java -Xmx16G -jar <pilon.jar>
to allocate 16GB to the JVM.
Usage: pilon --genome genome.fasta [--frags frags.bam] [--jumps jumps.bam] [--unpaired unpaired.bam]
[...other options...]
pilon --help for option details
INPUTS:
--genome genome.fasta
The input genome we are trying to improve, which must be the reference used
for the bam alignments. At least one of --frags or --jumps must also be given.
--frags frags.bam
A bam file consisting of fragment paired-end alignments, aligned to the --genome
argument using bwa or bowtie2. This argument may be specifed more than once.
--jumps jumps.bam
A bam file consisting of jump (mate pair) paired-end alignments, aligned to the
--genome argument using bwa or bowtie2. This argument may be specifed more than once.
--unpaired unpaired.bam
A bam file consisting of unpaired alignments, aligned to the --genome argument
using bwa or bowtie2. This argument may be specifed more than once.
--bam any.bam
A bam file of unknown type; Pilon will scan it and attempt to classify it as one
of the above bam types.
OUTPUTS:
--output prefix
Prefix for output files
--outdir directory
Use this directory for all output files.
--changes
If specified, a file listing changes in the <output>.fasta will be generated.
--vcf
If specified, a vcf file will be generated
--vcfqe
If specified, the VCF will contain a QE (quality-weighted evidence) field rather
than the default QP (quality-weighted percentage of evidence) field.
--tracks
This options will cause many track files (*.bed, *.wig) suitable for viewing in
a genome browser to be written.
CONTROL:
--variant
Sets up heuristics for variant calling, as opposed to assembly improvement;
equivalent to "--vcf --fix all,breaks".
--chunksize
Input FASTA elements larger than this will be processed in smaller pieces not to
exceed this size (default 10000000).
--diploid
Sample is from diploid organism; will eventually affect calling of heterozygous SNPs
--fix fixlist
A comma-separated list of categories of issues to try to fix:
"snps": try to fix individual base errors;
"indels": try to fix small indels;
"gaps": try to fill gaps;
"local": try to detect and fix local misassemblies;
"all": all of the above (default);
"bases": shorthand for "snps" and "indels" (for back compatibility);
"none": none of the above; new fasta file will not be written.
The following are experimental fix types:
"amb": fix ambiguous bases in fasta output (to most likely alternative);
"breaks": allow local reassembly to open new gaps (with "local");
"circles": try to close circlar elements when used with long corrected reads;
"novel": assemble novel sequence from unaligned non-jump reads.
--dumpreads
Dump reads for local re-assemblies.
--duplicates
Use reads marked as duplicates in the input BAMs (ignored by default).
--iupac
Output IUPAC ambiguous base codes in the output FASTA file when appropriate.
--nonpf
Use reads which failed sequencer quality filtering (ignored by default).
--targets targetlist
Only process the specified target(s). Targets are comma-separated, and each target
is a fasta element name optionally followed by a base range.
Example: "scaffold00001,scaffold00002:10000-20000" would result in processing all of
scaffold00001 and coordinates 10000-20000 of scaffold00002.
If "targetlist" is the name of a file, each line will be treated as a target
specification.
--threads
Degree of parallelism to use for certain processing (default 1). Experimental.
--verbose
More verbose output.
--debug
Debugging output (implies verbose).
--version
Print version string and exit.
HEURISTICS:
--defaultqual qual
Assumes bases are of this quality if quals are no present in input BAMs (default 15).
--flank nbases
Controls how much of the well-aligned reads will be used; this many bases at each
end of the good reads will be ignored (default 10).
--gapmargin
Closed gaps must be within this number of bases of true size to be closed (100000)
--K
Kmer size used by internal assembler (default 47).
--mindepth depth
Variants (snps and indels) will only be called if there is coverage of good pairs
at this depth or more; if this value is >= 1, it is an absolute depth, if it is a
fraction < 1, then minimum depth is computed by multiplying this value by the mean
coverage for the region, with a minumum value of 5 (default 0.1: min depth to call
is 10% of mean coverage or 5, whichever is greater).
--mingap
Minimum size for unclosed gaps (default 10)
--minmq
Minimum alignment mapping quality for a read to count in pileups (default 0)
--minqual
Minimum base quality to consider for pileups (default 0)
--nostrays
Skip making a pass through the input BAM files to identify stray pairs, that is,
those pairs in which both reads are aligned but not marked valid because they have
inconsistent orientation or separation. Identifying stray pairs can help fill gaps
and assemble larger insertions, especially of repeat content. However, doing so
sometimes consumes considerable memory.